Zinc-α2-glycoprotein 1 attenuates non-alcoholic fatty liver disease by negatively regulating tumour necrosis factor-α
2019-10-28TingLiuXinLuoZhengHongLiJunChengWuShengZhengLuoMingYiXu
Ting Liu, Xin Luo, Zheng-Hong Li, Jun-Cheng Wu, Sheng-Zheng Luo, Ming-Yi Xu
Abstract BACKGROUND Zinc-α2-glycoprotein 1 (AZGP1) plays important roles in metabolism-related diseases. The underlying molecular mechanisms and therapeutic effects of AZGP1 remain unknown in non-alcoholic fatty liver disease (NAFLD).AIM To explore the effects and potential mechanism of AZGP1 on NAFLD in vivo and in vitro.METHODS The expression of AZGP1 and its effects on hepatocytes were examined in NAFLD patients, CCl4-treated mice fed a high fat diet (HFD), and human LO2 cells.RESULTS AZGP1 levels were significantly decreased in liver tissues of NAFLD patients and mice. AZGP1 knockdown was found to activate inflammation; enhance steatogenesis, including promoting lipogenesis [sterol regulatory elementbinding protein (SREBP)-1c, liver X receptor (LXR), fatty acid synthase (FAS),acetyl-CoA carboxylase (ACC), and stearoyl CoA desaturase 1 (SCD)-1],increasing lipid transport and accumulation [fatty acid transport protein (FATP),carnitine palmitoyl transferase (CPT)-1A, and adiponectin], and reducing fatty acid β-oxidation [farnesoid X receptor (FXR) and peroxisome proliferatoractivated receptor (PPAR)-α]; accelerate proliferation; and reverse apoptosis in LO2 cells. AZGP1 overexpression (OV-AZGP1) had the opposite effects.Furthermore, AZGP1 alleviated NAFLD by blocking TNF-α-mediated inflammation and intracellular lipid deposition, promoting proliferation, and inhibiting apoptosis in LO2 cells. Finally, treatment with OV-AZGP1 plasmid dramatically improved liver injury and eliminated liver fat in NAFLD mice.CONCLUSION AZGP1 attenuates NAFLD with regard to ameliorating inflammation,accelerating lipolysis, promoting proliferation, and reducing apoptosis by negatively regulating TNF-α. AZGP1 is suggested to be a novel promising therapeutic target for NAFLD.
Key words: Non-alcoholic fatty liver disease; Zinc-α2-glycoprotein 1; Tumour necrosis factor-α; Inflammation; Lipid metabolism
INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) affects 25% of the global adult population and is highly prevalent on all continents. The highest rates are reported in South America and the Middle East (31%-32%), followed by Asia (27%), the USA, and Europe (23%-24%)[1-3]. The contribution of NAFLD to the burden of end-stage liver disease and hepatocellular carcinoma (HCC) is growing and is projected to surpass hepatitis C as the principal indication for liver transplantation[4]. Given the high prevalence and the fact that there is no effective treatment, a better understanding of the pathogenesis is essential to identify targets and overcome this epidemic disease.
Zinc-α2-glycoprotein (AZGP1) was first discovered in human plasma and subsequently purified by Burgi in 1961[5]. Later, it was found in many body fluids,such as sweat, saliva, and urine[5,6]. AZGP1, a 41 kDa glycoprotein assigned to chromosome 7q22.1, is a secreted glycoprotein that is synthesized by epithelial cells and adipocytes of many organs[7]. AZGP1 plays a role in lipolysis, regulation of metabolism, and cell proliferation and differentiation[7]. AZGP1 could also be a potential marker for the diagnosis of obesity-induced NAFLD[8-10]. Furthermore,AZGP1 was found to be correlated with tumour necrosis factor-α (TNF-α) in the adipose tissue and liver of obese mice[11]. However, the complex molecular mechanisms underlying AZGP1 and obesity-induced NAFLD are not fully understood. Our study focused on how AZGP1 regulates inflammation and lipid metabolism; clarified the functions of AZGP1 in NAFLD patients, LO2 cells, and NAFLD mice; and provided new insights for future therapeutic strategies against NAFLD.
MATERIALS AND METHODS
Human liver tissues
Liver tissues were obtained from patients with chronic hepatitis B (CHB, n = 6) or NAFLD (n = 6) who received percutaneous liver needle biopsy. The diagnosis of NAFLD was based on the detection of hepatic steatosis (intrahepatic triglyceride content ≥ 5.5% by magnetic resonance imaging or steatosis in ≥ 5% of hepatocytes by histology) and exclusion of the patients who consumed > 30 g of alcohol per day and other liver diseases by histology and suitable investigations[12-14]. Our research focused on hepatocytes in NAFLD patients, so there were no associated hepatitis or toxic substances use. All liver samples were classified as steatotic according to NAFLD activity scoring[15]and the NASH Clinical Research Network scoring system[16];inflammation grade and fibrosis stage were based on Scheuer's classification[17]. CHB group samples were identified as inflammation grade (G0-2)[17]/fibrosis stage (S0-1)[17]/steatosis degree (F0-1)[15,16], and NAFLD group samples were identified as G0-2[17]/S0-1[17]/F3[15,16]. Six liver samples (CHB and NAFLD groups, n = 3 each) were subjected to immunohistochemistry (IHC); another six samples (CHB and NAFLD groups, n = 3 each) were subjected to quantitative polymerase chain reaction (qPCR)and Western blot analysis. All patients provided written informed consent, and the study was approved by the Ethics Committee of Shanghai General Hospital.
NAFLD mouse model
Male C57BL/6 mice were randomly separated into four groups [control (low fat diet,LFD), HFD (high fat diet), HFD + CCl4, and HFD + CCl4+ AZGP1; n = 6 each] at 8 wk of age (Shanghai SLAC Laboratory Animal Co. Ltd, Shanghai, China). All mice were fed an LFD or HFD (containing 60% calories from fat, BioServ, Frenchtown, NJ,United States) for 14 wk and then sacrificed. Mice were injected with CCl4(Sigma-Aldrich Corp, St. Louis, MO, United States) three times a week during the final 6 weeks to establish a liver injury and cirrhosis model in the HFD + CCl4and HFD +CCl4+ AZGP1 groups; while oil (2 μL/g, 3 times a week) was used for the LFD and HFD groups. A plasmid overexpressing AZGP1 (OV-AZGP1) was intravenously injected into the tail of mice once every 2 d during the final 2 wk[18,19]in the HFD +CCl4+ AZGP1 group, and other groups were injected with a control plasmid (GV315).The plasmid GV315 or OV-AZGP1 for mice (25 μg, Shanghai GenePharma Co. Ltd,Shanghai, China, Supplementary Table 1) was mixed with Lipofectamine 2000 (40 μL,Invitrogen Life Technologies, CA, United States) and incubated at room temperature for 30 min. Blood samples and liver tissues were collected and stored at -80 °C. All samples of the four groups were subjected to haematoxylin and eosin (HE) staining,Sirius red staining, qPCR, Western blot, immunofluorescence (IF) staining,measurements of serum alanine transaminase (ALT), and oil red O (ORO) staining.Animal experiments were carried out according to the guidelines of Shanghai Jiao Tong University School of Medicine and approved by the Ethics Committee of Shanghai General Hospital.
Cell culture and plasmid transfection
A human normal hepatic cell line (LO2) was used in this study. Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (HyClone, United States)supplemented with 100 mg/ml streptomycin, 100 IU/ml penicillin, and 10% foetal bovine serum (HyClone). Overexpression was accomplished by transfection with the plasmids pEX-3 and OV-AZGP1 for 48 h (Shanghai GenePharma Co. Ltd; Supplementary Table 1). Knockdown was accomplished by transfection with the pGPU6 plasmid AZGP1 shRNA (sh-AZGP1) and TNF-α shRNA (sh-TNF-α) for 48 h(Shanghai GenePharma Co. Ltd; Supplementary Table 1). Transfection was performed using Lipofectamine 2000 according to the manufacturer's instructions. Cells were treated with 300 μmol/L palmitic acid (PA, Sigma-Aldrich Corp) under serum-free conditions.
qPCR
Hieff qPCR SYBR Green Master Mix (Applied Biosystems, CA, United States) and Hieff First Strand cDNA Synthesis Super Mix for RT-qPCR (Applied Biosystems)were used for qPCR. The mRNA levels of specific genes were normalized to that of βactin. The primers used are listed in Supplementary Table 2.
Western blot analysis
Western blot was performed using the following antibodies: Anti-TNF-α (1:100); antiinterleukin (IL)-1β and anti-nuclear factor-κ-gene binding (NF-κB) p50 (1:200); anti-Bcell lymphoma 2 (Bcl2) (1:500); anti-AZGP1, anti-IL-6, anti-liver X receptor (LXR),anti-fatty acid synthase (FAS), anti-stearoyl CoA desaturase 1 (SCD-1), anti-farnesoid X receptor (FXR), anti-carnitine palmitoyl transferase (CPT)-1A, anti-fatty acid transport protein (FATP), anti-proliferating cell nuclear antigen (PCNA), anti-cyclin D1, and anti-caspase 3 (1:1000); and horseradish peroxidase (HRP)-conjugated glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:10000); the secondary antibody was HRP-immunoglobulin G (IgG, 1:20000). Detailed information on the primary antibodies is provided in Supplementary Table 3.
IHC and IF staining
Antibodies for AZGP1 (1:200), TNF-α (1:20), IL-6 (1:200), and NF-κB (1:50) were used.Cell nuclei were stained with 4'6-diamidino-2-phenylindole (DAPI) for IF staining.The secondary antibody used for IF staining was Alexa Fluor 488/594 AffiniPure goat anti-rabbit IgG (H+L) (Santa Cruz Biotechnology, TX, United States). IHC images were obtained using an inverted microscope (Leica Microsystems, Germany), and IF images were obtained using a laser confocal microscope (Leica Microsystems).
Cell proliferation assay
Cell Counting Kit-8 (CCK-8) assays (Roche Applied Science, IN, United States) were used to examine cell proliferation. The optical density (OD) value was measured with a microplate reader (Leica Microsystems) at 450 nm.
Cell apoptosis analysis
Cells were washed with phosphate buffer solution (PBS) and resuspended in 100 μL of binding buffer containing 5 μL of Annexin V-FITC and 10 μL of propidium iodide(PI) staining solution. After incubation for 10 minutes, the samples were analysed using a flow cytometer (BD AccuriTMC6, ThermoFisher Scientific, MA, United States).
Cell cycle analysis
Cells were harvested, washed with PBS, and fixed in 70% ice-cold ethanol overnight.Then, the cells were washed with PBS and stained with PI (50 ng/mL) in staining buffer supplemented with RNase A (50 mg/mL) at room temperature in the dark for 30 min. Analyses of the cell cycle distribution were conducted with an Accuri C6 flow cytometer (BD Biosciences, United States) and ModFit LT software.
ORO staining
Lipid accumulation was evaluated by ORO staining. Briefly, samples were fixed with 4% paraformaldehyde for 30 min and then stained with ORO (Yeasen Bio., Beijing,China) for 15 min. Then, the cells were destained with 60% isopropanol. After washing in PBS repeatedly, the cells were examined by light microscopy to determine lipid droplet accumulation.
Measurements of serum ALT
Serum ALT concentrations were determined using an ALT Activity Assay Kit(Nanjing Jiancheng Bioengineering Institute, Jiangsu, China). The OD value was measured with a microplate reader (Leica Microsystems) at 510 nm.
HE and Sirius red staining
Formalin-fixed mouse liver tissue samples were embedded in paraffin, cut into 5-μm thick sections, and then stained with HE and Sirius red. Cell nuclei were stained with hematoxylin for counterstaining. The images were obtained using an inverted microscope (Leica Microsystems, Germany).
Statistical analysis
The results are expressed as the mean ± standard deviation (SD), and all experiments were repeated at least three times. Comparisons between two groups were analyzed using an independent t-test; differences among groups were performed by one-way ANOVA. All statistical analyses were done using SPSS 19.0 (SPSS Inc., Chicago, IL,United States). A P-value less than 0.05 was considered statistically significant.
RESULTS
AZGP1 is markedly down-regulated in liver samples of NAFLD patients
To understand the expression and function of AZGP1 in NAFLD, we first detected AZGP1 in human NAFLD samples. IHC staining revealed that compared with the CHB patients, the liver tissues from NAFLD patients had only a few AZGP1-positive cells (Figure 1A). Furthermore, AZGP1 levels were significantly lower in the NAFLD livers than in the CHB livers at both the mRNA and protein levels (Figure 1B and C).Therefore, we speculate that the loss of AZGP1 is an unknown pathogenic factor in human NAFLD.
AZGP1 negatively regulates liver inflammatory factors in human hepatocytes
Inflammatory factors such as TNF-α, IL-1β, IL-6, NF-κB, and MCP-1 were reported to be significantly elevated in mice fed an HFD compared to mice fed an LFD[20-22]. We examined the effects of AZGP1 on liver inflammation by overexpressing or knocking down AZGP1 in LO2 cells (Supplementary Figure 1) after exposure to PA. PA treatment elevated the expression of inflammatory factors such as TNF-α, IL-1β, IL-6,NF-κB, and MCP-1 at both the mRNA (Figure 2A and D) and protein levels (except MCP-1, Figure 2B and E). OV-AZGP1 led to a dramatic decrease in these inflammatory factors at the mRNA and protein levels (Figure 2A and B); in contrast,sh-AZGP1 led to a significant increase in these factors (Figure 2D and E) with or without PA. Furthermore, IF assays showed that PA suppressed AZGP1 and activated TNF-α protein in LO2 cells (Figure 2C and F). OV-AZGP1 decreased and sh-AZGP1 enhanced TNF-α protein expression (Figure 2C and F) regardless of incubation with PA. Double-label fluorescence revealed that AZGP1 and TNF-α co-localized in nucleoplasmic foci and identified that their expression was negatively correlated(Figure 2C and F). Therefore, our findings suggested that AZGP1 negatively regulates liver inflammatory factors, especially TNF-α, in PA-stimulated LO2 cells.
AZGP1 suppresses lipid synthesis and accelerates lipolysis in human hepatocytes
Lipotoxicity in hepatocytes induced by PA has been implicated in the development of NAFLD. ORO staining showed that exposure to PA led to strong intracellular lipid accumulation in LO2 cells (Figure 3A). Cellular fat droplets dramatically decreased in OV-AZGP1 ± PA cells compared to the control cells (Figure 3A). On the other hand,markedly increased fat droplets were observed in the sh-AZGP1 ± PA groups compared to the control group (Figure 3B). Then, we examined whether AZGP1 affects lipid metabolism pathways in hepatocytes. First, lipogenesis factors, including metabolic nuclear receptors (SREBP-1c and LXR) and lipogenic enzymes [FAS, acetyl-CoA carboxylase (ACC) and SCD-1] in the liver, were checked. OV-AZGP1 caused an apparent decrease in SREBP-1c, LXR, FAS, ACC, and SCD-1 mRNA and protein levels in LO2 cells incubated with PA or not, when compared to each control cell (Figure 3C and E). Additionally, compared to the controls, sh-AZGP1 led to a more obvious increase (Figure 3D and F). FXR and peroxisome proliferator-activated receptor(PPAR)-α are nuclear receptors that mediate fatty acid β-oxidation in hepatocytes.OV-AZGP1 increased the FXR and PPAR-α mRNA and protein expression levels that were suppressed by PA (Figure 3C and E). In contrast, sh-AZGP1 decreased their expression (Figure 3D and F). Then, the expression of FATP and CPT-1A (which are related to lipid transport and fatty acid β-oxidation) in LO2 cells was detected. Only after incubation with PA, OV-AZGP1 decreased FATP or increased CPT-1A expression (Figure 3C and E); sh-AZGP1 reversed these effects (Figure 3D and F).Adiponectin is an important adipokine that suppresses lipid accumulation in the liver[23]. Expression of adiponectin mRNA decreased in PA-treated LO2 cells (Figure 3C). OV-AZGP1 significantly increased adiponectin mRNA levels while sh-AZGP1 markedly decreased adiponectin mRNA levels in LO2 cells (Figure 3C and D). In brief, AZGP1 suppressed lipid biogenesis, transport, and accumulation, and accelerated fatty acid β-oxidation in PA-stimulated LO2 cells.
AZGP1 promotes proliferation and inhibits apoptosis in human hepatocytes
Next, we clarified the effects of AZGP1 on the proliferation and apoptosis of LO2 cells. PA treatment inhibited the expression of PCNA and cyclin D1 at both the mRNA and protein levels (Figure 4A, B, E and F); OV-AZGP1 increased those levels(Figure 4A and B), and sh-AZGP1 decreased them (Figure 4E and F) in both low(without PA) and high (with PA) fat environments. CCK-8 assays also indicated that PA treatment suppressed cell proliferation; OV-AZGP1 significantly reversed this effect, and sh-AZGP1 further suppressed proliferation in both low and high fat environments (Figure 4C and G). Flow cytometry also proved that OV-AZGP1 promoted cell proliferation (pEX-3: 0.30 vs OV-AZGP1: 0.42, P = 2.00E-04; pEX-3+PA:0.27 vs OV-AZGP1+PA: 0.38, P = 9.00E-04) and that AZGP1 deficiency inhibited cell proliferation (sh-CTRL: 0.43 vs sh-AZGP1: 0.36, P = 1.20E-03; sh-CTRL + PA: 0.33 vs sh-AZGP1 + PA: 0.27, P = 2.00E-05; Figure 4D and H) under both low and high fat conditions. Furthermore, we found that the percentage of cells in the G0/G1 phase decreased, and the percentage of cells in the S and G2/M phases increased in OVAZGP1 LO2 cells compared to pEX-3 cells with or without PA stimulation; sh-AZGP1 had the opposite effect (Figure 4D and H). Additionally, the effect of AZGP1 on cell apoptosis was investigated in LO2 cells. PA treatment inhibited the expression of Bcl2 and elevated the expression of caspase 3 at both the mRNA and protein levels (Figure 4I, J, L and M); as expected, OV-AZGP1 counteracted these effects (Figure 4I and J)and sh-AZGP1 enhanced them (Figure 4L-M). The results of Annexin V-PI staining showed that the proportion of early-stage apoptotic cells significantly increased following exposure to PA (Figure 4K and N). Furthermore, OV-AZGP1 decreased the percentage of apoptotic cells (pEX-3: 26% vs OV-AZGP1: 9%, P = 1.80E-03; pEX-3 +PA: 30% vs OV-AZGP1 + PA: 21%, P = 2.30E-03), and sh-AZGP1 increased the percentage of apoptotic cells (sh-CTRL: 12% vs shAZGP1: 16%, P = 1.56E-02; sh-CTRL+ PA: 15% vs shAZGP1 + PA: 23%, P = 3.00E-04; Figure 4K and N). Therefore, AZGP1 accelerated proliferation and reduced apoptosis in PA-stimulated LO2 cells.
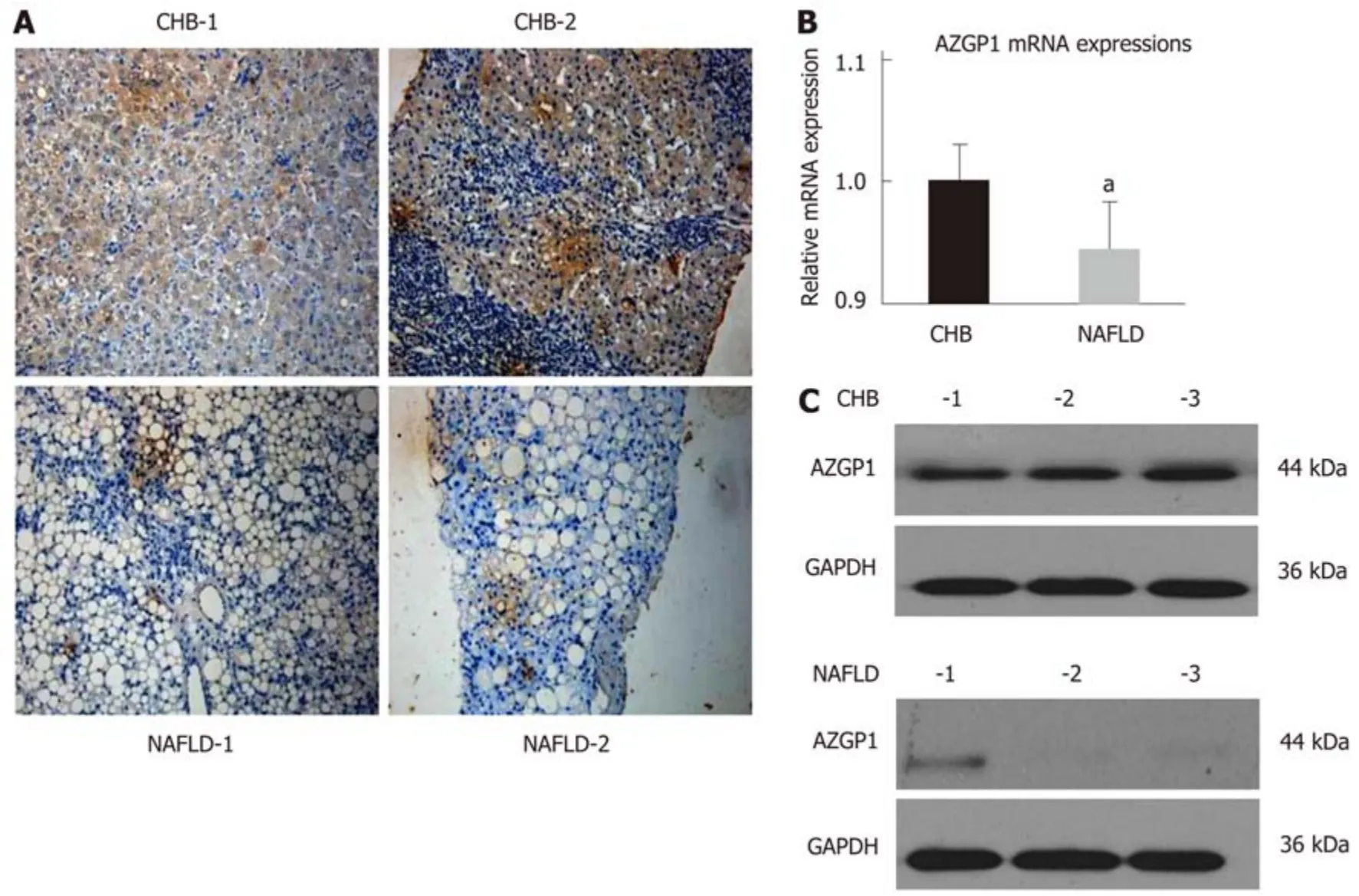
Figure 1 Expression of AZGP1 in liver tissues of chronic hepatitis B and non-alcoholic fatty liver disease patients. IHC-stained AZGP1 is showed in human liver tissues (A) of two chronic hepatitis B (CHB) and non-alcoholic fatty liver disease (NAFLD) patients. AZGP1 mRNA (B) and protein (C) expression is showed in liver tissues of another group of CHB and NAFLD patients (n = 3 each). aP < 0.05 vs CHB group. CHB: Chronic hepatitis B; NAFLD: Non-alcoholic fatty liver disease.
AZGP1 exerts multiple functions by blocking TNF-α in human hepatocytes
To further explore the modulatory effect of TNF-α on AZGP1, we constructed OVTNF-α in OV-AZGP1 LO2 cells. OV-TNF-α could reverse the effects of OV-AZGP1 that down-regulated inflammatory factors NF-κB and IL-6 (Figs. 5A-B). More fat droplets accumulated in the OV-AZGP1 + OV-TNF-α group than in the OV-AZGP1 group (Figure 5C). OV-TNF-α could down-regulate pro-proliferative factors PCNA and cyclin D1 which were up-regulated by OV-AZGP1 (Figure 5A and B). OV-TNF-α significantly reversed the pro-proliferative effect of OV-AZGP1 (Figure 5D). And OVTNF-α reversed the effect on apoptotic factors Bcl2 and caspase 3 in OV-AZGP1 cells(Figure 5A and B). OV-TNF-α visibly increased the percentage of apoptotic cells,which decreased in OV-AZGP1 cells (OV-AZGP1/OV-AZGP1 + OV-TNFα: 0.09/0.17,P = 5.09E-03; Figure 5E). Then, we treated LO2 cells with sh-AZGP1 or sh-TNF-α. Sh-TNF-α also reversed the effects of sh-AZGP1 on NF-κB and IL-6 (Figure 5F and G).Fewer cellular fat droplets were seen in the sh-AZGP1 + sh-TNF-α group than in the sh-AZGP1 group (Figure 5H). The sh-TNF-α cells were unable to inhibit PCNA and cyclin D1 in the sh-AZGP1 cells (Figure 5F and G). Sh-TNF-α significantly reversed the proliferation-inhibiting effect of sh-AZGP1 (Figure 5I). The changes of Bcl2 and caspase 3 in sh-AZGP1 cells were reversed by sh-TNF-α (Figure 5F and G). Also sh-TNF-α visibly reduced the percentage of apoptotic cells, which was elevated by sh-AZGP1 (sh-AZGP1/sh-AZGP1 + sh-TNFα: 0.15/0.10, P = 1.59E-03; Figure 5J). We concluded that AZGP1 cannot inactivate inflammation, reduce lipid accumulation,promote proliferation, or suppress apoptosis without TNF-α in LO2 cells.
AZGP1 negatively regulates TNF-α, resulting in the alleviation of liver steatosis and fibrosis in NAFLD mice
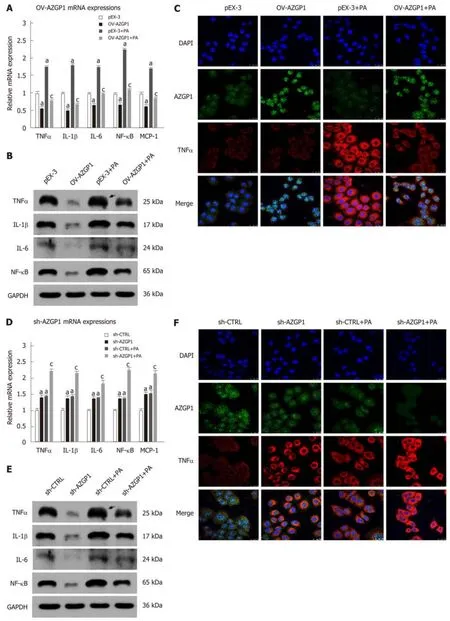
Figure 2 Inhibition of inflammation by AZGP1 in LO2 cells. TNF-α, IL-1β, IL-6, NF-κB, and MCP-1 mRNA and protein expression was detected in LO2 cells, pEX-3 ± PA/OV-AZGP1 ± PA groups (A-B), and sh-CTRL ± PA/sh-AZGP1 ± PA groups (D-E). IF staining of LO2 cells shows nuclei (blue), AZGP1 protein (green), and TNF-α protein (red) in these groups (C/F). aP < 0.05 vs pEX-3 or sh-CTRL cells; aP < 0.05 vs pEX-3 + PA or sh-CTRL + PA cells.
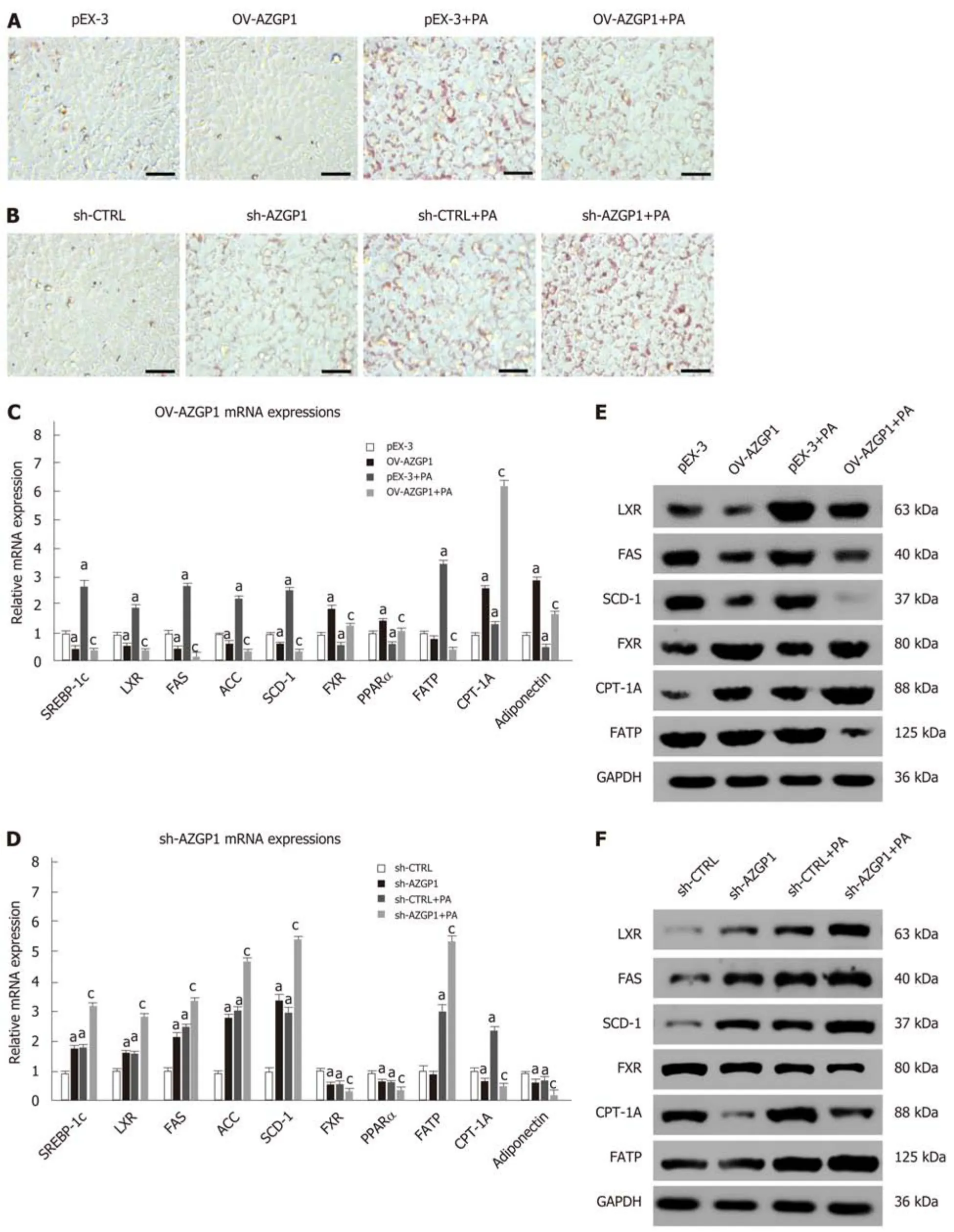
Figure 3 Regulation of lipid metabolism by AZGP1 in LO2 cells. ORO staining of lipids in LO2 cells is shown in pEX-3 ± PA/OV-AZGP1 ± PA groups (A) and sh-CTRL ± PA/sh-AZGP1 ± PA groups (B). SREBP-1c, LXR, FAS, ACC, SCD-1, FXR, PPAR-α, FATP, CPT-1A, and adiponectin mRNA and protein expression was detected in the pEX-3 ± PA/OV-AZGP1 ± PA groups (C/E) and sh-CTRL ± PA/sh-AZGP1 ± PA groups (D/F). aP < 0.05 vs pEX-3 or sh-CTRL cells; cP < 0.05 vs pEX-3 + PA or sh-CTRL + PA cells.
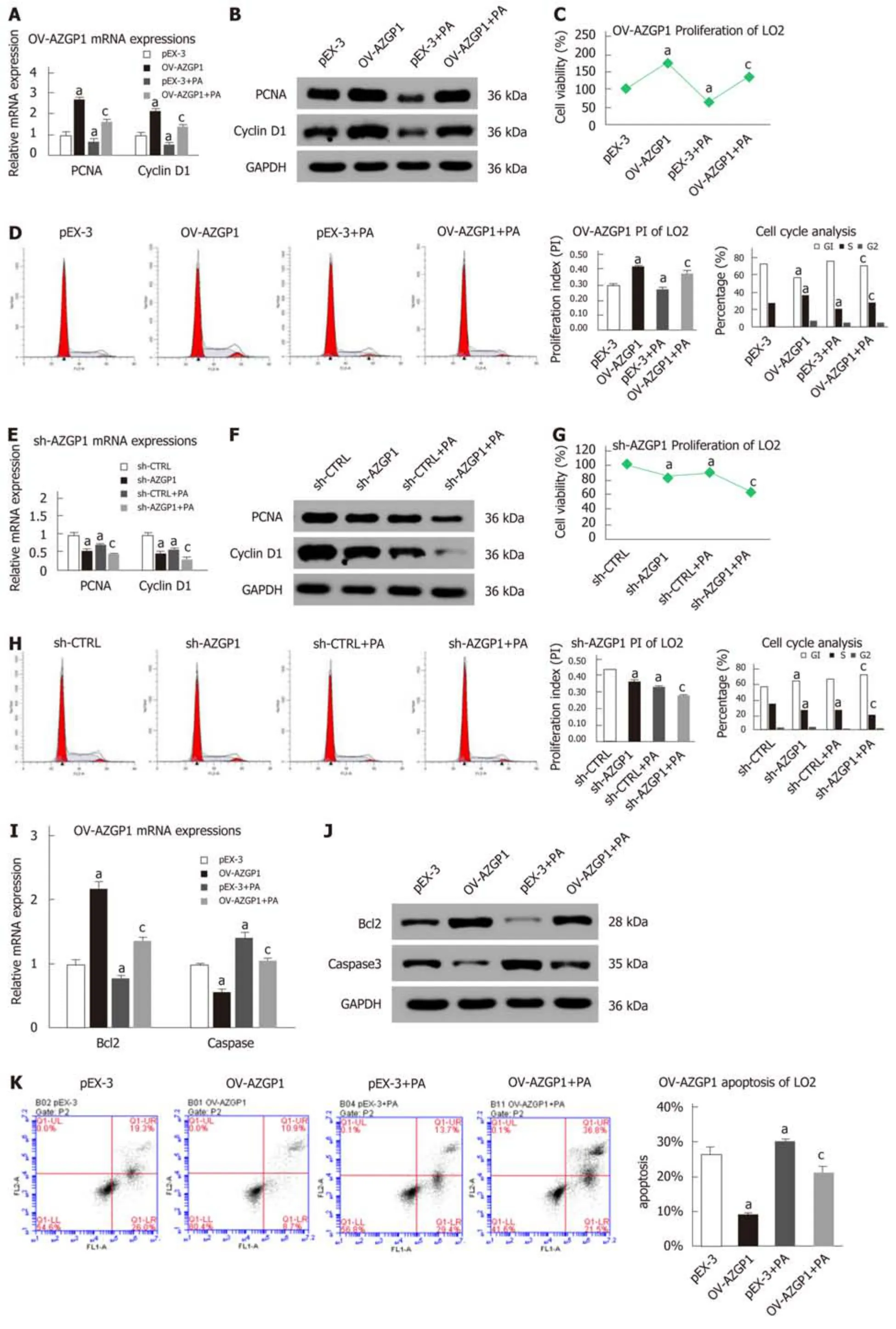
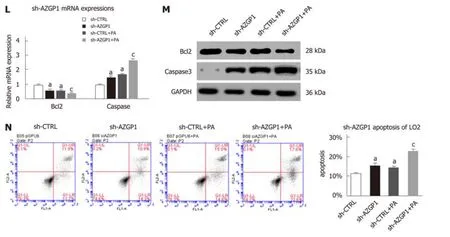
Figure 4 Proliferation and apoptosis regulation by AZGP1 in LO2 cells. PCNA and cyclin D1 mRNA (A/E) and protein (B/F) expression was detected in LO2 cells. Cell viability was determined using CCK-8 assays (C/G). Cell cycle and proliferation analyses were conducted by flow cytometry (D/H). Bcl2 and caspase 3 mRNA (I/L) and protein (J/M) expression was detected in LO2 cells. The rate of apoptosis was tested using flow cytometry (K/N). aP < 0.05 vs pEX-3 or sh-CTRL cells;cP < 0.05 vs pEX-3 + PA or sh-CTRL + PA cells.
To explore the role of AZGP1 in NAFLD, we established HFD-induced liver steatosis and CCl4-induced liver fibrosis mouse models; furthermore, the OV-AZGP1 plasmid was injected as a therapy. HE staining showed that mice in the HFD group developed steatosis; additionally, hepatocytes stored fat droplets, especially those hepatocytes located around the central vein. Mice in the HFD + CCl4group showed lobular inflammation together with hepatocellular ballooning; furthermore, AZGP1 treatment significantly alleviated hepatic steatosis and inflammation in the HFD + CCl4+AZGP1 group (Figure 6A). Sirius red staining revealed that there was no fibrosis in the mice of the control or HFD groups and there was advanced fibrosis in the HFD +CCl4group; furthermore, AZGP1 treatment slightly ameliorated the degree of liver fibrosis in the HFD + CCl4+ AZGP1 group (Figure 6B). Hepatic AZGP1 mRNA and protein levels were lower in the HFD ± CCl4groups than in the control group, while these levels were highest in the HFD + CCl4+ AZGP1 group with OV-AZGP1 plasmid injection (Figure 6C-E). In contrast, hepatic TNF-α mRNA and protein levels were notably higher in the HFD ± CCl4groups than in the controls; AZGP1 treatment significantly down-regulated these levels (Figure 6C-E). Moreover, double IF staining for AZGP1 and TNF-α in hepatocytes indicated that the two proteins co-localized in nucleoplasmic foci and AZGP1 displayed a negative correlation with TNF-α (Figure 6E). Overall, AZGP1 counteracted TNF-α, relieving liver steatosis or fibrosis in HFD and CCl4-induced NAFLD mice.
AZGP1 ameliorates liver inflammation and lipid deposition in NAFLD mice
Serum ALT levels markedly increased in the HFD ± CCl4groups compared with the control group, but they obviously decreased in the HFD + CCl4+ AZGP1 group compared with the HFD + CCl4group (Figure 6F). IL-1β, IL-6, NF-κB, and MCP-1 significantly increased in the HFD ± CCl4groups compared with the controls, but AZGP1 treatment reversed the increase (Figure 6G and H). ORO staining indicated mild diffuse steatosis in the control mice and aggravated steatosis in the HFD ± CCl4groups; AZGP1 treatment obviously attenuated hepatic steatosis caused by HFD and CCl4(Figure 6I). SREBP-1c, LXR, FAS, ACC, SCD-1, and FATP were notably upregulated in the HFD ± CCl4groups compared to the control group; similarly, AZGP1 treatment reversed these effects (Figure 6J and K). Interestingly, CPT-1A was further up-regulated in the HFD + CCl4+ AZGP1 group compared to the HFD + CCl4group(Figure 6J and K). Moreover, FXR, PPARα, and adiponectin were extraordinarily down-regulated in the HFD ± CCl4groups compared with the control group, and AZGP1 treatment reversed this effect (Figure 6J and K). In conclusion, AZGP1 regulated inflammation and lipid metabolism factors, resulting in the amelioration of HFD- and CCl4-induced NAFLD in mice.
DISCUSSION
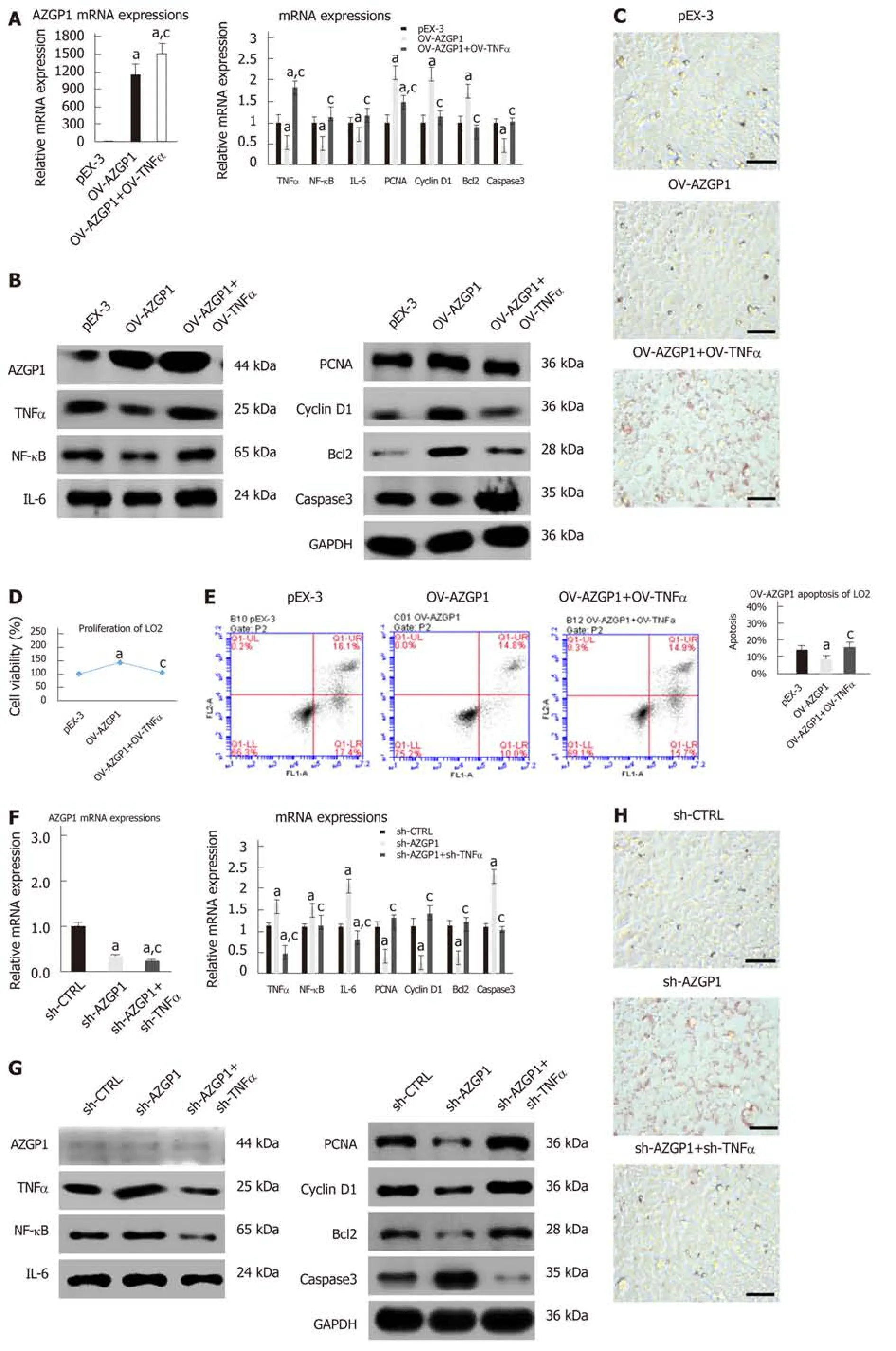
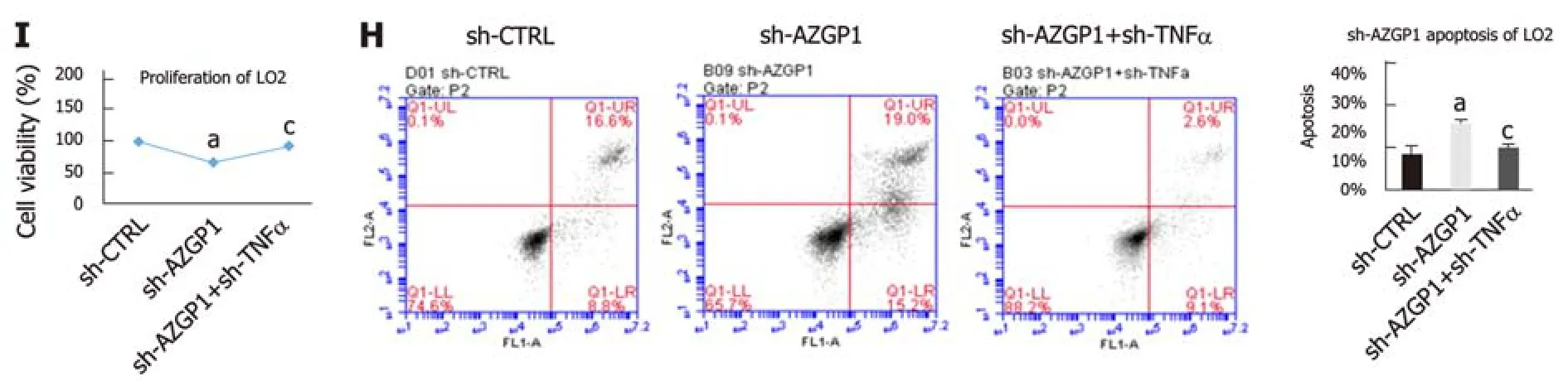
Figure 5 AZGP1 exerts multiple functions by blocking TNF-α in LO2 cells. All experiments were performed in LO2 cells, which were divided into six groups (pEX-3, OV-AZGP1, OV-AZGP1 + OV-TNF-α, sh-CTRL, sh-AZGP1, and sh-AZGP1 + sh-TNF-α). AZGP1, TNF-α, NF-κB, IL-6, PCNA, cyclin D1, Bcl2, and caspase 3 mRNA (A, F) and protein (B, G) expression was detected. ORO staining (C, H) of LO2 cells is shown. Cell viability (D, I) was determined using CCK-8 assays. The rate of apoptotic LO2 cells was tested using flow cytometry (E, J). aP < 0.05 vs pEX-3 or sh-CTRL cells; cP < 0.05 vs OV-AZGP1 or sh-AZGP1 cells.
NAFLD has already become the most common cause of chronic liver disease in many parts of the world[24]. Over the last decade, the clinical burden of NAFLD has not only been limited to liver-related morbidity and mortality, but there has also been increasing evidence that NAFLD is a multisystem disease that influences extrahepatic organs and regulatory pathways[25]. Therefore, it is necessary to explore the pathogenesis and treatment of NAFLD. Liu et al found that AZGP1 dramatically decreased with obesity in Chinese people and HFD-induced obese mice[26]. In our study, we found that the expression of AZGP1 was significantly low in the liver tissues of NAFLD patients and mice (HFD + CCl4model) at both the mRNA and protein levels. Therefore, we inferred that the loss of AZGP1 in the liver highly suggested the progression of NAFLD.
Inflammation is conducive to the pathogenesis of the majority of acute and chronic liver diseases, such as NAFLD, alcoholic fatty liver disease, liver transplantation, and viral hepatitis[27,28]. The inflammatory responses could be characterized by a common spectrum of genes and endogenous mediators involved, including inflammatory cytokines such as TNF-α, IL-1β, IL-6, and IL-8[29,30]. Li et al[20]found that an HFD,compared to an LFD, resulted in a dramatic increase in the serum levels of TNF-α, IL-1β, IL-6, and ALT. In our study, AZGP1 down-regulated inflammatory factors (TNFα, IL-1β, IL-6, NF-κB, and MCP-1) in human hepatocytes and NAFLD mice. Knocking down AZGP1 had the opposite effect on inflammation in hepatocytes. Furthermore,our earlier study indicated that the loss of AZGP1 was related to cell proliferation and apoptosis[31]. Our data suggested that AZGP1 could promote proliferation and inhibit apoptosis, while the loss of AZGP1 had the opposite effects in hepatocytes.
Although there is strong evidence linking AZGP1 with lipolysis[32-34], the explicit physiological function of AZGP1 in liver lipid metabolism in vitro remains unclear.Our research illuminated the significance of AZGP1 in hepatic lipid metabolism related to NAFLD. Knockdown of AZGP1 increased the deposition of lipids; on the contrary, the overexpression of AZGP1 reversed this effect in hepatocytes and the liver of NAFLD mice under low or high fat conditions. It could be concluded that AZGP1 plays an important role in hepatic lipid degradation both in vitro or in vivo.Hepatic lipid accumulation stems from an imbalance between lipid usability (from circulating lipid uptake or de novo fatty acid synthesis) and lipid disposal (via triglyceride-rich lipoprotein secretion or free fatty acid oxidation). Thus, we detected the key enzymes in these pathways to evaluate the influence of AZGP1 on these processes. FAS is a rate-limiting enzyme in de novo fatty acid synthesis[35]. ACC is a rate-limiting enzyme in the synthesis of malonyl-CoA (a key precursor for fatty acid biological synthesis)[36]. SCD-1 is a microsomal enzyme that is important for the synthesis of monounsaturated fatty acids[37]. Our study indicated that AZGP1 inhibited the activities of these enzymes (FAS, ACC, and SCD-1), which increased hepatic intracellular lipid deposition. Down-regulation of lipogenesis after the overexpression of AZGP1 indicated that AZGP1 was an important negative regulator of adipogenesis in hepatocytes and mice. Liver metabolic nuclear receptors play significant roles in the pathogenesis of NAFLD[38]. The receptors are involved in lipid uptake, storage, oxidation, and export and lipolysis. PPARα and FXR (the bile acidactivated nuclear receptor) take part in lipogenesis[39]; SREBP-1c and LXR(transcription factors) positively regulate fatty acid synthesis in hepatocytes[40]. Our research also indicated that AZGP1 promoted lipolysis and inhibited lipogenesis by regulating the metabolic nuclear receptors PPARα, FXR, SREBP-1c, and LXR in hepatocytes and mice. Furthermore, we also found that AZGP1 down-regulated FATP to decrease lipid transport and up-regulated CPT-1A to promote fatty acid βoxidation in high fat conditions. Finally, adiponectin is reported to activate fatty acid oxidation by up-regulating CPT-1A, to suppress lipogenesis by down-regulating FAS and ACC, and to prevent free fatty acid influx by decreasing CD36, thereby relieving fatty liver[41]. We also observed that AZGP1 may indirectly suppress lipid accumulation by activating adiponectin in hepatocytes. Therefore, our results indicate new molecular mechanisms for metabolic nuclear receptors in NAFLD pathogenesis and provide novel targets for the advancement of treatments for NAFLD.
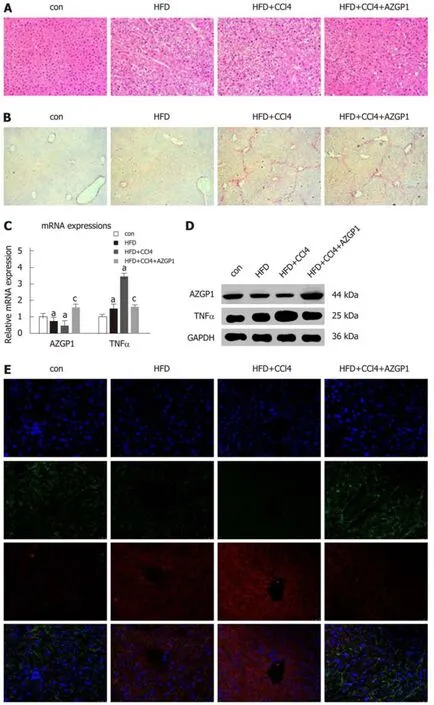
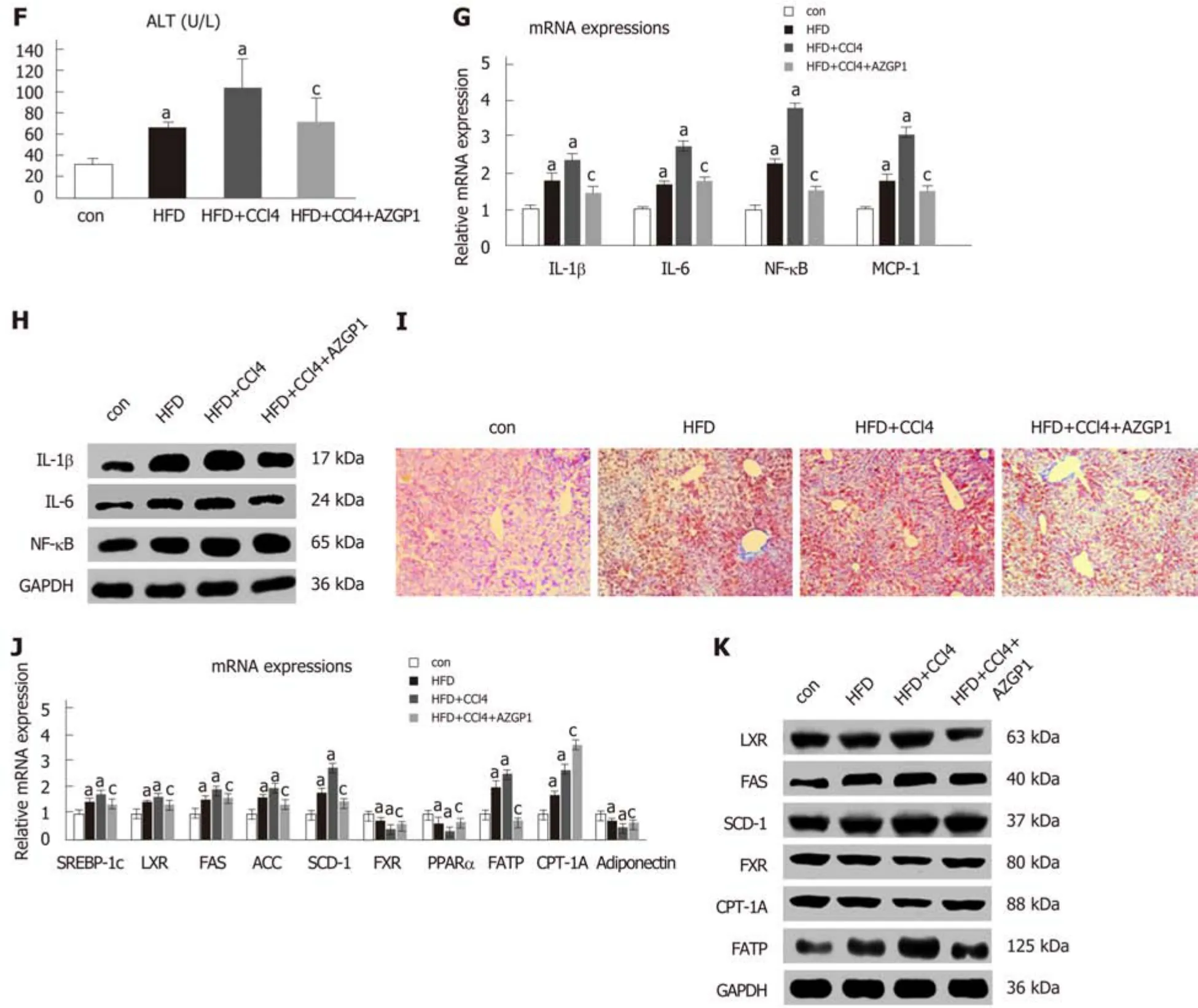
Figure 6 Expression of AZGP1/TNF-α, inflammation, and lipid metabolism regulation by AZGP1 in the non-alcoholic fatty liver disease mouse model. Liver tissue images after HE (A) and Sirius red (B) staining are shown in four mouse groups [control (con), HFD, HFD + CCl4, and HFD + CCl4 + AZGP1]. Levels of AZGP1 and TNF-α mRNA (C) and protein (D) were detected in the four mouse groups. IF staining (E) with DAPI (blue) and for AZGP1 (green) and TNF-α (red) was also examined in mice. Serum ALT levels (F) were examined using an ELISA kit in the four mouse groups. IL-1β, IL-6, NF-κB, and MCP-1 mRNA (G) and protein (H)expression levels were detected in the four groups. Representative images of ORO staining (I) are shown for the four groups. SREBP-1c, LXR, FAS, ACC, SCD-1,FXR, PPARα, FATP, CPT-1A, and adiponectin mRNA (J) and protein (K) expression was detected in the four groups. aP < 0.05 vs control mice; cP < 0.05 vs HFD +CCl4 mice.
Finally, we characterized the signalling pathway modulated by AZGP1 in hepatocytes. Mracek et al[11]discovered that the inhibitory effect of TNF-α would lead to a reduction in AZGP1 and its production, thus increasing susceptibility to lipid accumulation in adipose tissue and obese mice. TNF-α is an important activator of NF-κB, which, in turn, is a potent inducer of TNF-α; this positive feedback is key in chronic inflammatory conditions[42]. We found that sh-AZGP1 could not activate hepatocyte inflammation (NF-κB or IL-6), apoptosis, and intracellular lipid accumulation or inhibit proliferation when used with sh-TNF-α simultaneously. Also,OV-TNF-α could partially reverse the effect of OV-AZGP1. Double IF staining for AZGP1 and TNF-α in LO2 cells showed a negative relation between AZGP1 and TNFα. Therefore, we concluded that the lack of AZGP1 played a role in exacerbating NAFLD by activating TNF-α.
Regarding the effect of AZGP1, our previous study also found that the deletion of AZGP1 induced epithelial-mesenchymal transition (EMT), accompanied by confusion of energy metabolism, reduction of cell proliferation and apoptosis, and increased invasion, and finally, AZGP1 suppressed hepatic carcinogenesis by blocking TGFβ1-extracellular regulated protein kinases (ERK2) signaling pathway[31]. A study by Hyun Sik et al found that AZGP1 aggravated the development of inflammatory diseases by forming a vicious circle with IL-17, such as rheumatoid arthritis patients with an HFD[43]. It has also been found that AZGP1 inhibited the inflammatory response associated with obesity by blocking the NF-κB signaling pathway[44]. Moreover,AZGP1 reduced insulin resistance by promoting the insulin receptor substrate-1 (IRS-1)/protein kinase B (AKT) signaling pathway[44]. Research on AZGP1 and fatty liver found that AZGP1 reduced intracellular lipid deposition by up-regulating lipolysis genes and down-regulating the expression of adipogenic genes[44]; Xiao et al[8]also found that AZGP1 can significantly inhibit lipogenesis, promote lipolysis and fatty acid β oxidation, and reduce PA-induced intracellular lipid deposition. Our results are similar to these studies.
Since the role of AZGP1 remains unclear, there is the possibility that AZGP1 loss could be a consequence of an unknown cause and not a cause of damage. Our study found that the expression of AZGP1 was reduced in both NAFLD patients and CCl4-treated mice fed an HFD, which are similar to previous studies[8,11,45]. We found that the disease state of NAFLD was aggravated after knocking down AZGP1 by artificially applying plasmid-based shRNA to LO2 cells. However, there was no relevant literature on the exact cause of AZGP1 loss. Although the specific mechanism is still unclear, the expression of AZGP1 could be partially attributed to the acetylation of histone, which regulated gene viability by altering the structure of chromatin[46]. Tian et al[47]reported that deacetylation of histone H4 decreased AZGP1 expression by inhibiting the transcription factor Ikaros binds to the promoter of AZGP1 in HCC cells. Daniel and his colleagues found that AZGP1 is upregulated in lung adenocarcinoma due to acetylation[48]. Kong et al[49]had also reported that AZGP1 is absent from pancreatic ductal adenocarcinoma due to histone deacetylation.However, whether histone deacetylation contributes to the decreased expression of AZGP1 found in our study requires further investigation. If we want to study whether the loss of AZGP1 alone will directly induce the occurrence of injury, we may need AZGP1 knockout mice, molecular biology studies and so on.
In conclusion, AZGP1 loss aggravates liver inflammation, promotes intracellular lipid accumulation, suppresses lipid degradation metabolism, reduces cell proliferation, and promotes cell apoptosis. AZGP1 reverses these effects and attenuates NAFLD by blocking TNF-α. We propose that AZGP1 may be a promising therapeutic candidate for NAFLD.
ARTICLE HIGHLIGHTS
Research background
Non-alcoholic fatty liver disease (NAFLD) is increasingly threatening people's health. Zinc-α2-glycoprotein 1 (AZGP1) was originally considered as a potential tumor marker, but subsequent studies have shown that it is also expressed in the liver, heart, lungs, and prostate. AZGP1 plays an important role in metabolism-related diseases, but the specific pathogenesis and therapeutic effects of AZGP1 in NAFLD remain uncertain.
Research motivation
Our findings will provide a basis for the application of AZGP1 in the therapy of NAFLD.
Research objectives
To examine the expression of AZGP1 in NAFLD patients, CCl4-treated mice fed a high fat diet(HFD), and human LO2 cells, and explore biological functions and potential mechanisms of AZGP1 in NAFLD.
Research methods
We detected the expression of AZGP1 in the liver tissues of NAFLD patients and CCl4-treated mice fed an HFD and human LO2 cells by qPCR and Western blot. The effects of AZGP1 on hepatocytes in vitro and in vivo were then assessed by overexpression and knockdown of AZGP1 in human LO2 cells and in an NAFLD mouse model. In further molecular mechanism research,LO2 cells were treated with sh-AZGP1 or combination of sh-AZGP1 and sh-TNF-α, OV-AZGP1 or combination of OV-AZGP1, and OV-TNF-α to investigate the specific effects of AZGP1 in vitro.
Research results
In the current study, we found that AZGP1 was significantly decreased in liver tissues from both humans and mice. Loss of AZGP1 activated inflammation, enhanced steatogenesis, increased lipid transport and accumulation, decreased fatty acid β-oxidation, promoted proliferation, and inhibited apoptosis in human LO2 cells. Over-expression of AZGP1 played an opposite role. In addition, AZGP1 attenuated NAFLD by blocking TNF-α-mediated inflammation and intracellular lipid deposition, promoting proliferation, and inhibiting apoptosis in LO2 cells.Finally, treatment with OV-AZGP1 plasmid in NAFLD mice significantly ameliorated liver damage and eliminated liver fat.
Research conclusions
AZGP1 is down-regulated in NAFLD, which could inhibit inflammation, accelerate lipolysis,accelerate proliferation, and reduce apoptosis via suppressing TNF-α. AZGP1 exerts a protective role against NAFLD.
Research perspectives
This study provides new insight into the role of AZGP1 in relieving NAFLD by down-relating TNF-α. AZGP1 might be a potential therapeutic approach to prevent and treat NAFLD.
杂志排行

World Journal of Gastroenterology的其它文章
- Chinese guidelines on management of hepatic encephalopathy in cirrhosis
- Sexual health and fertility for individuals with inflammatory bowel disease
- High mobility group box-1 release from H2O2-injured hepatocytes due to sirt1 functional inhibition
- Clostridium butyricum alleviates intestinal low-grade inflammation in TNBS-induced irritable bowel syndrome in mice by regulating functional status of lamina propria dendritic cells
- CARMA3/NF-κB signaling contributes to tumorigenesis of hepatocellular carcinoma and is inhibited by sodium aescinate
- Laparoscopy-assisted pylorus-preserving gastrectomy for early gastric cancer: A retrospective study of longterm functional outcomes and quality of life