Role of ion channels in gastrointestinal cancer
2019-10-28KyleAndersonRobertCormierPatriciaScott
Kyle J Anderson, Robert T Cormier, Patricia M Scott
Abstract In their seminal papers Hanahan and Weinberg described oncogenic processes a normal cell undergoes to be transformed into a cancer cell. The functions of ion channels in the gastrointestinal (GI) tract influence a variety of cellular processes,many of which overlap with these hallmarks of cancer. In this review we focus on the roles of the calcium (Ca2+), sodium (Na+), potassium (K+), chloride (Cl-) and zinc (Zn2+) transporters in GI cancer, with a special emphasis on the roles of the KCNQ1 K+ channel and CFTR Cl- channel in colorectal cancer (CRC). Ca2+ is a ubiquitous second messenger, serving as a signaling molecule for a variety of cellular processes such as control of the cell cycle, apoptosis, and migration.Various members of the TRP superfamily, including TRPM8, TRPM7, TRPM6 and TRPM2, have been implicated in GI cancers, especially through overexpression in pancreatic adenocarcinomas and down-regulation in colon cancer. Voltage-gated sodium channels (VGSCs) are classically associated with the initiation and conduction of action potentials in electrically excitable cells such as neurons and muscle cells. The VGSC NaV1.5 is abundantly expressed in human colorectal CRC cell lines as well as being highly expressed in primary CRC samples. Studies have demonstrated that conductance through NaV1.5 contributes significantly to CRC cell invasiveness and cancer progression. Zn2+transporters of the ZIP/SLC39A and ZnT/SLC30A families are dysregulated in all major GI organ cancers, in particular, ZIP4 up-regulation in pancreatic cancer(PC). More than 70 K+ channel genes, clustered in four families, are found expressed in the GI tract, where they regulate a range of cellular processes,including gastrin secretion in the stomach and anion secretion and fluid balance in the intestinal tract. Several distinct types of K+ channels are found dysregulated in the GI tract. Notable are hERG1 upregulation in PC, gastric cancer (GC) and CRC, leading to enhanced cancer angiogenesis and invasion, and KCNQ1 down-regulation in CRC, where KCNQ1 expression is associated with enhanced disease-free survival in stage II, III, and IV disease. Cl- channels are critical for a range of cellular and tissue processes in the GI tract, especially fluid balance in the colon. Most notable is CFTR, whose deficiency leads to mucus blockage, microbial dysbiosis and inflammation in the intestinal tract. CFTR is a tumor suppressor in several GI cancers. Cystic fibrosis patients are at a significant risk for CRC and low levels of CFTR expression are associated with poor overall disease-free survival in sporadic CRC. Two other classes of chloride channels that are dysregulated in GI cancers are the chloride intracellular channels (CLIC1, 3 &4) and the chloride channel accessory proteins (CLCA1,2,4). CLIC1 & 4 are upregulated in PC, GC, gallbladder cancer, and CRC, while the CLCA proteins have been reported to be down-regulated in CRC. In summary, it is clear, from the diverse influences of ion channels, that their aberrant expression and/or activity can contribute to malignant transformation and tumor progression.Further, because ion channels are often localized to the plasma membrane and subject to multiple layers of regulation, they represent promising clinical targets for therapeutic intervention including the repurposing of current drugs.
Key words: Ion channels; Gastrointestinal cancer; Colorectal cancer; Gastric cancer;Pancreatic cancer; Esophageal cancer; Hepatocellular carcinoma; Prognostic biomarker;Novel therapies; Clinical targets
INTRODUCTION
In 2019, the American Cancer Society estimates that there will be more than 1.76 million new cases of cancer in the United States, accompanied by more than 607,000 cancer deaths[1]. Of these, the digestive system will have the highest number of new cases, and the second highest number of cancer deaths. The lifespan of cells in the gastrointestinal (GI) tract is very short. Propagated from stem cells, the epithelial cells of the stomach, small intestine, and colorectum are typically replaced in a matter of days and are some of the most replicative tissues in the body. This turnover is necessary due to the constant physical, chemical, and biological insults these tissues endure. This rapid proliferation increases the likelihood of cells in these tissues acquiring and accumulating oncogenic mutations.
The basic functions of the GI epithelium are: (1) to act as a physical barrier that selectively allows for the absorption of nutrients; while (2) excluding toxic or pathogenic substances; and (3) to excrete substances to aid in the digestion process.These functions require large quantities of water, ions, and nutrients to be transported across the epithelial layer. The significant driving force for this work is achieved through the use of ion gradients.
The unequal distribution of ions is required for the survival and function of any cell. This includes everything from concentration gradients across cellular and organellar membranes to gradients within the cytosol from one end to the other of a polarized cell. The distribution of ions is a consequence of the localization and activation of a variety of ion-specific channels, co-transporters, and pumps. Ion channels typically have a gating mechanism controlling when they are open or closed,and allow for the passive movement of select ions down their concentration gradient.In contrast to this passive dissipation of gradients, pumps make use of ATP hydrolysis to actively set up gradients. Co-transporters, or secondary pumps, exploit the energy gained by moving one ion down its gradient to power the movement of another ion or molecule against its gradient. Precise regulation of these elements in response to changing environmental conditions and the subsequent changes in ion concentration or flux are necessary for a multitude of cellular processes including proliferation, motility, absorption/secretion, apoptosis and many others. The goal of this review will be to summarize what we know about the role of ion channel dysfunction as it pertains to cancer development within the GI epithelium. We focus on the four main classes of ion channels: potassium (K+), chloride (Cl-), sodium (Na+),and calcium (Ca2+) and zinc (Zn2+) transporters and the major epithelial cancers that arise in the GI tract: colorectal cancer (CRC), gastric cancer (GC), esophageal cancer(EC), pancreatic cancer (PC) and hepatocellular carcinoma (HCC). For other important GI transporters and aquaporins and other types of cancers we refer readers to other specific reviews.
Nearly 20 years ago, Hanahan and Weinberg presented certain criteria that a normal cell must acquire to be transformed into a cancer cell[2]. These hallmarks of cancer have been expanded upon since that seminal paper, but their basic principles still remain[3]. The functions of ion channels influence a variety of cellular processes,many of which overlap heavily with these hallmarks of cancer. For this reason, cancer has been described as a channelopathy[4], with a recent review by Prevarskaya et al[5]asking whether cancer hallmarks are primarily oncochannelopathies. For example,Ca2+channels play major roles in the control of cellular growth and proliferation, as well as the control of cell death[6]. K+and Cl-channels are essential for the localized swelling and shrinking of different areas of a cell, necessary for cell migration[7].Separate from their function as channels, studies of protein interactions with various ion channels have demonstrated their involvement in diverse processes such as cytoskeletal architecture and protein targeting[8]. It is clear, from the diverse influences of ion channels, that their aberrant expression and/or dysfunction can contribute to the transformation of normal cells into malignancy[4-5,9-15]. As discussed by Djamgoz et al[12]ion channels are expressed and dysregulated in all cancers throughout the multistage process, from initiation to metastasis. This is certainly true for the GI tract.
POTASSIUM CHANNELS
K+channels play a major role in maintenance of plasma membrane (PM) potential.The action of the Na+/K+-ATPase transporter, H+/K+-ATPase transporter, and the NKCC cotransporter, coupled with the exit of K+ions from the cell down their electrochemical gradient maintains a net intracellular negative charge at the PM. This hyperpolarized membrane potential is then used to drive the active transport of various molecules against their gradient. This is especially important in the GI epithelium, which must continuously transport mass quantities of water, electrolytes,and nutrients. With 77 genes coding for K+channels, they are the largest, most diverse group of ion channels in the human genome. In addition, many of these genes have known splice variants, can be post-translationally modified, or form complexes with regulatory subunits. This immense variability in K+channel function highlights the importance of being able to fine-tune K+conductance and brings into question what other functions these channels might be serving. Generally, K+channels are classified as either voltage-gated, Ca2+-activated, inward rectifier channels, or 2P-domain channels.
Whereas Navchannels are classically associated with the rapid depolarization of excitable cells, voltage-gated K+(Kv) channels are responsible for re-polarization during an action potential. In non-excitable cells, such as those of the GI epithelium,the typical role of these channels is hyperpolarization of the PM. This negative membrane potential facilitates Ca2+signaling and is required for regulation of intracellular pH and cell volume. Owing to these broad influences, K+channels are implicated in a variety of cellular and tissue functions including cell proliferation and differentiation, pigmentation, hearing and the mammalian cochlea, contractility,circadian rhythms, migration, wound healing, cell cycle progression, apoptosis,autophagy, metabolism, angiogenesis, stem cell dynamics, and carcinogenesis,including cancer cell proliferation, invasion, migration and metastasis[9,11,16-20]. Notably,the mechanisms underlying loss of control of K+channels are still not well understood[18].
KCNQ1
Very prominent among the Kvchannels contributing to cancer risk is KCNQ1, which demonstrates flexibility in gating permitting functional versatility[21]. In the GI tract KCNQ1 assembles with the β-subunits KCNE2 (gastric) or KCNE3 (intestinal)converting it from a voltage-dependent channel in the stomach into a voltageindependent, constitutively open channel in the intestine[22]. In gastric parietal cells,luminal KCNQ1/KCNE2 is essential for gastric acid secretion, working in conjunction with a H+/K+-ATPase. In the intestinal and colonic crypts, KCNQ1/KCNE3 is located basolaterally, and establishes the driving force for cAMP-mediated Cl-secretion through CFTR, necessary for mucus hydration[23,24]. In the colon, blocking the activity of KCNQ1/KCNE3 nearly entirely abolishes cAMP-mediated Cl-secretion, versus only about 50% in the small intestine, demonstrating a reliance on KCNQ1 in the colon[25].
KCNQ1 deficiency in humans and animal models
Humans carrying germline mutations in KCNQ1 (Jervell and Lange-Nielsen and Romano-Ward syndromes) develop a range of pathologies, most notably cardiac arrhythmia (long and short QT), but also hearing loss, elevated gastrin levels, gastric hyperplasia and in some cases gastric neoplasia[26-30]. These phenotypes are well modeled in Kcnq1 knockout mice that develop inner ear defects, imbalance, chronic gastritis, gastric hyperplasia, and gastric metaplasia[31,32].
KCNQ1 and GI cancer
There is strong evidence for KCNQ1 functioning as a tumor suppressor in GI cancers.The first data came from Sleeping Beauty (SB) transposon mutagenesis screens for intestinal cancer in mice. Kcnq1 was the third-ranked common insertion site (CIS)gene (just behind Apc and Rspo2, well-known Wnt/β-catenin pathway signaling genes), with a predicted loss of function and 14 unique mutation sites, among 77 CIS genes identified in the first SB screen published in Science in 2009[33]. Kcnq1 was then identified as a CIS gene in three subsequent SB screens for intestinal cancer[34-36].Kcnq1's role was then confirmed in crosses of Kcnq1 knockout mice to the ApcMinmouse model of intestinal cancer where Kcnq1-deficiency significantly enhanced tumor phenotypes, including the development of invasive adenocarcinomas[37]. The role of KCNQ1 in human CRC was then investigated, finding that maintenance of KCNQ1 mRNA and protein levels was associated with significant disease-free and overall survival in stage II, III, and IV CRC[37,38]. Notably, for stage IV CRC patients following hepatic resections, maintenance of KCNQ1 protein expression conferred a 23-month survival advantage[37]. In other GI cancers Kcnq1 was a CIS gene in two SB screens for PC[39,40], one SB screen for HCC[41]and in one SB screen for GC, with a predicted loss of function[42]. Additional evidence in GC is provided by the phenotype of Kcnq1 knockout mice that develop gastric hyperplasia, metaplasia and occasional neoplasia[31,32]and in studies of human gastric cells where treatment of cells with atrial natriuretic peptide reduced cell proliferation by upregulating KCNQ1 expression[43].In studies of HCC in human tissue and HCC cell lines, expression of KCNQ1 was down-regulated by promoter hypermethylation associated with epithelial to mesenchymal transition (EMT), and poor patient prognosis[44]. Additionally, in HCC it was reported that KCNQ1 regulated and sequestered β-catenin via physical interactions at the PM[44].
Although KCNQ1 deficiency is associated with poor outcome in CRC[37,38,45]and in HCC[44], the mechanisms underlying tumor suppression are not well understood.However, one clue is that KCNQ1 is localized to the base of the intestinal epithelial crypt which is the site of the stem cell compartment and the likely site of origin of CRC[46]. Functional significance of crypt localization was demonstrated by Than et al[37]who found that crypts isolated from KCNQ1-deficient colon epithelium displayed increased clonogenicity suggesting a possible selective advantage for tumor development. In addition, several studies demonstrate involvement of KCNQ1 with the Wnt/β-catenin pathway[38,44,45,47]. The Wnt/β-catenin pathway is vitally important in intestinal epithelial physiology and pathophysiology, with deregulation of the pathway contributing to over 80% of CRCs as well as a large percentage of HCCs. An early study in Xenopus oocytes demonstrated that β-catenin up regulated KCNQ1-mediated currents by promoting its insertion into PM without any effect on transcription[47]. More recent studies have looked at the interactions of KCNQ1 and βcatenin specifically in GI cancers. Analysis of 386 human stage II and III CRC tumors found correlation between KCNQ1 membrane-associated protein and nuclear βcatenin protein expression, which was surprising as KCNQ1-low expression was associated with poor outcome[38]. It was proposed that KCNQ1 may be downregulated by promoter methylation in some cancers so that in the presence of βcatenin, as seen in most CRC, KCNQ1 would be upregulated, but if in addition the promoter becomes methylated KCNQ1 would be down-regulated. In contrast, a second study found that β-catenin directly negatively regulated KCNQ1 transcription in several CRC cell lines[45]. This group also found that KCNQ1 promoted cell membrane localization of β-catenin. This had the effect of limiting oncogenesis both by preventing nuclear localization of β-catenin and by maintaining adherens junctions that prevent EMT. A third study reported that in HCC cell lines, KCNQ1 expression was enhanced by treatment with a methyltransferase inhibiter suggesting that expression may be down-regulated by promoter methylation[44]. However, KCNQ1 appeared to sequester β-catenin at the cell membrane and to limit its transcriptional activity. The situation is further complicated by the lncRNA KCNQ1OT1, which through the recruitment of chromatin and DNA modifying proteins, silences multiple genes in the KCNQ1 region[48]. KCNQ1OT1 has been associated with poor patient survival in several GI cancers, including CRC[49], EC[50], and HCC[51]. KCNQ1OT1 itself is regulated by β-catenin. The Kugoh group has demonstrated that nuclear β-catenin activates the transcription of KCNQ1OT1 through a TCF-1 binding site within its promoter region[52]. These studies suggest that multiple factors affect the interactions between KCNQ1 and β-catenin, and that the balance of these factors differs among cancers. But apparently loss of KCNQ1 activity, through whatever means, can provide a selective advantage to the tumor, as each of these studies demonstrates that KCNQ1 is a tumor suppressor. Channel openers such as retigabine have been developed as treatments for diseases caused by KCNQ1-deficiency[53]. Thus, understanding the contribution of KCNQ1 to cancer progression could lead to new cancer therapeutic opportunities.
KCNE2 and KCNE3
Given its role as the β-subunit in the KCNQ1/KCNE2 channel in gastric tissue it is not surprising that deficiency for KCNE2 contributes to human GC cell growth and progression. This was supported by studies in Kcne2 knockout mice that develop gastritis cystica profunda and neoplasia, pyloric polyadenomas and invasive adenocarcinomas, linked to upregulation of cyclin D1[54-58]. Kcne3 (intestinal β-subunit)knockout mice also partially phenocopied loss of KCNQ1, demonstrating a disruption in intestinal Cl-transport[46].
Human ether-a-go-go related gene 1
While KCNQ1 seems to have tumor-suppressive effects, another Kvchannel, the human ether-a-go-go related gene 1 (hERG1) channel, has been implicated as an oncogene in various GI cancers including CRC[59-62], PC[62-66], EC[67-70], and GC[68,71-74]. In CRC, while hERG1 is not expressed in normal colonic mucosa, a distinct upregulation in expression is reported in adenocarcinomas, with the highest levels occurring in metastatic cancers, where hERG1 expression was associated with poor patient prognosis, and with little to no expression in adenomas. hERG1 expression levels and activity were positively correlated to cell migration using channel inhibitors and clones expressing various levels of the protein[59]. This role in cancer cell migration and invasion was later expanded upon with the discovery of hERG1's interactions with β1-integrins in PM complexes. hERG1 was shown to modulate β1-integrin mediated VEGF-A secretion through the recruitment of PI3K and AKT[72]. In PC hERG1 overexpression was reported in 59% of tumors[66]where it promoted cancer cell migration via modulation of F-actin organization[64]. hERG1 expression was also associated with lymph node involvement, tumor grade, TNM stage and poor patient prognosis[66].One report linked hERG1 to dysregulation of the EGFR signaling pathway. In EC,hERG1 expression was associated with progression from Barrett's esophagus (BE) to EC, again associated with poor patient prognosis[70]. Finally, in GC, hERG1 overexpression was reported in 69% of cancers, where it promoted angiogenesis by mediating VEGF-A secretion via AKT-dependent regulation of HIF-1, and similarly,its expression was associated with poor patient prognosis[71-74].
EAG1
Another ether-a-go-go family member, EAG1, is also reported to function as an oncogene in several GI cancers, including CRC[18,75,76], EC[75], and HCC[16,62,75,77]. In all cases EAG1 is reported to be over-expressed, and associated with invasion of cancer cells and poor patient prognosis.
Other K+ channels
At least nine other K+channels have been reported to be dysregulated in GI cancers.Those acting as oncogenes include KCNA5 in GC[73,78-80], and CRC[79]; KCNC1 in CRC[76]; KCND1 in GC[81]; KCNJ3 in PC[65,82]; KCNN4 in CRC[5,14,83], PC[65,84], and HCC;KCNS3 in CRC[85]; and KCNK9[5,86,87]in CRC. Those acting as potential tumor suppressors include KCNA3 in CRC[4,76,88]and PC[65,82,89], and KCNH5 in EC[90]. Of note,the pathophysiological actions of many of these channels seem to involve mechanisms that are independent of their channel action. An example is KCNS3/Kv9.3, an electrically silent subunit in excitable cells, whose expression has been found increased in CRC and lung cancer cells, with a knockdown showing interference at the G0/G1 and G1/S cell cycle transitions[85]. Overall, the great majority of K+channels discussed here function as oncogenes (with KCNQ1 a prominent exception),consistent with a model of K+channel dysregulation/overexpression being necessary for cancer cell cycle and tumor progression. See Table 1 for a full listing of potassium ion channels and their role in GI cancers.
CHLORIDE CHANNELS
The functions of Cl-channels in the GI tract include regulation of transepithelial transport (in particular major anions), volume control (osmoregulation), membrane potentials, lipid homeostasis, cell polarity, glucose and other metabolism, oxidative stress, inflammation, mucus, alterations in the microbiome, pH, cell motility,autophagy, mitochondrial dysfunction, apoptosis, cell polarity, cell-cell contact, stem cell function, and cellular immune responses[5,91-105]. All of these functions can be dysregulated in and contribute to malignant transformation, particularly in the GI tract due to its constant exposure to environmental influences.
CFTR
CFTR encodes a Cl-, HCO3-anion channel found primarily on the apical surfaces of luminal epithelial cells. Mutations in CFTR are the cause of the hereditary life shortening disease cystic fibrosis (CF). Recently CFTR has been shown to be a tumor suppressor in CRC and CFTR deficiency has been implicated in several other cancers.Because the causal connection between CFTR and cancer has been most strongly established for CRC, this section of the review will focus primarily on CRC.
Normal functions of CFTR
The CFTR gene found on chromosome 7 encodes an mRNA of 6128 nucleotides[106].The CFTR protein consists of two symmetrical halves. Each half contains 6 membrane spanning domains joined by an intracellular regulatory region. The membrane spanning domains assemble to form an aqueous pore that allows flow of Cl-and HCO3-ions down their electrochemical gradients. In the intestine the flow of ions is from the cytoplasm to lumen. Ion specificity is provided by the amino acids lining the pore. Opening and closing is regulated by binding of ATP to two nucleotide binding domains in the regulatory region. ATP binding is mediated by cAMP activation of PKA which phosphorylates a site to open up the ATP binding domains. In vivo cAMP activity is commonly regulated by cholinergic stimuli. Outflow of ions creates osmotic pressure for the flow of water in the same direction so that CFTR indirectly determines the flow of water as well[107,108]. CFTR also regulates other ion channels(Na+, K+, Ca2+, and other Cl-channels). For example, CFTR indirectly regulates the cellular ionic environment by inhibiting activity of the Na+importing channel SCNN1,which has the effect of further encouraging outflow of water and also by supporting additional HCO3-transporters[109]. CFTR also interacts with other membrane proteins to maintain epithelial tight junctions and barriers to fluid flow, adjusts the levels of acidity in secretions, and participates in the transport of sphingosine-1 phosphate, a regulator of cell adhesion and a signaling molecule for inflammation[99]. CFTR also contains a cytoplasmic C-terminal PDZ-binding motif. This domain interacts with PDZ-containing proteins involved in regulating the actin cytoskeleton and intracellular signaling[110,111]. CFTR is expressed on the apical surface of the intestinal epithelium throughout the length of the intestine. In the small intestine CFTR expression decreases from duodenum to ileum with strongest expression in the crypts[112]. In addition, functional CFTR is found on the brush border of villus cells and rare CFTR-high expressing cells throughout the small intestine[113]. In the colon CFTR is expressed in a proximal to distal gradient with highest concentration in the proximal region and cecum. CFTR is most highly expressed at the base of the crypt where intestinal epithelial stem cells reside[112,114]. Overall, CFTR is a major determinant of ion and water homeostasis and because it is highly expressed at the base of the crypt it is in a position to influence the intestinal stem cell compartment.
Cystic fibrosis
Inactivating germline mutations in CFTR cause the hereditary life shortening disease CF. CF is the most common autosomal recessive hereditary disease among Caucasians[115]. The most severe clinical manifestations are pulmonary inflammation and obstruction leading ultimately to pulmonary failure[116]. However, CFTR is expressed in many extra-pulmonary tissues including the linings of the pancreatic and biliary ducts where its loss ultimately leads to CF-related diabetes and CF-relatedliver disease[117,118]. CFTR dysfunction in the exocrine pancreas results in ion transport defects, β-islet cell-related disorders such as dysregulation of insulin release,obstruction of the pancreatic duct, chronic inflammation and cancer initiation[99,119]. CF patients also demonstrate defects in the male reproductive system, chronic sinusitis,and kidney stones. Older CF patients have dysregulation of glucose metabolism and sleep disorders caused by disruption of circadian rhythms[99]. CFTR is also highly expressed in the intestinal epithelium. Loss of CFTR in the intestine causes intestinal obstruction in the ileum and proximal colon known as meconium ileus in infants and distal intestinal obstruction in older patients. CF results in impaired absorption of nutrients due to pancreatic enzyme deficiency and possibly defects in lipid absorption[120]. CF patients are also prone to celiac disease, an intestinal disorder caused by gluten-mediated triggering of TH1 immune and antibody responses[102].These and other clinical manifestations of CF demonstrate the profound importance of CFTR activity in many tissues.
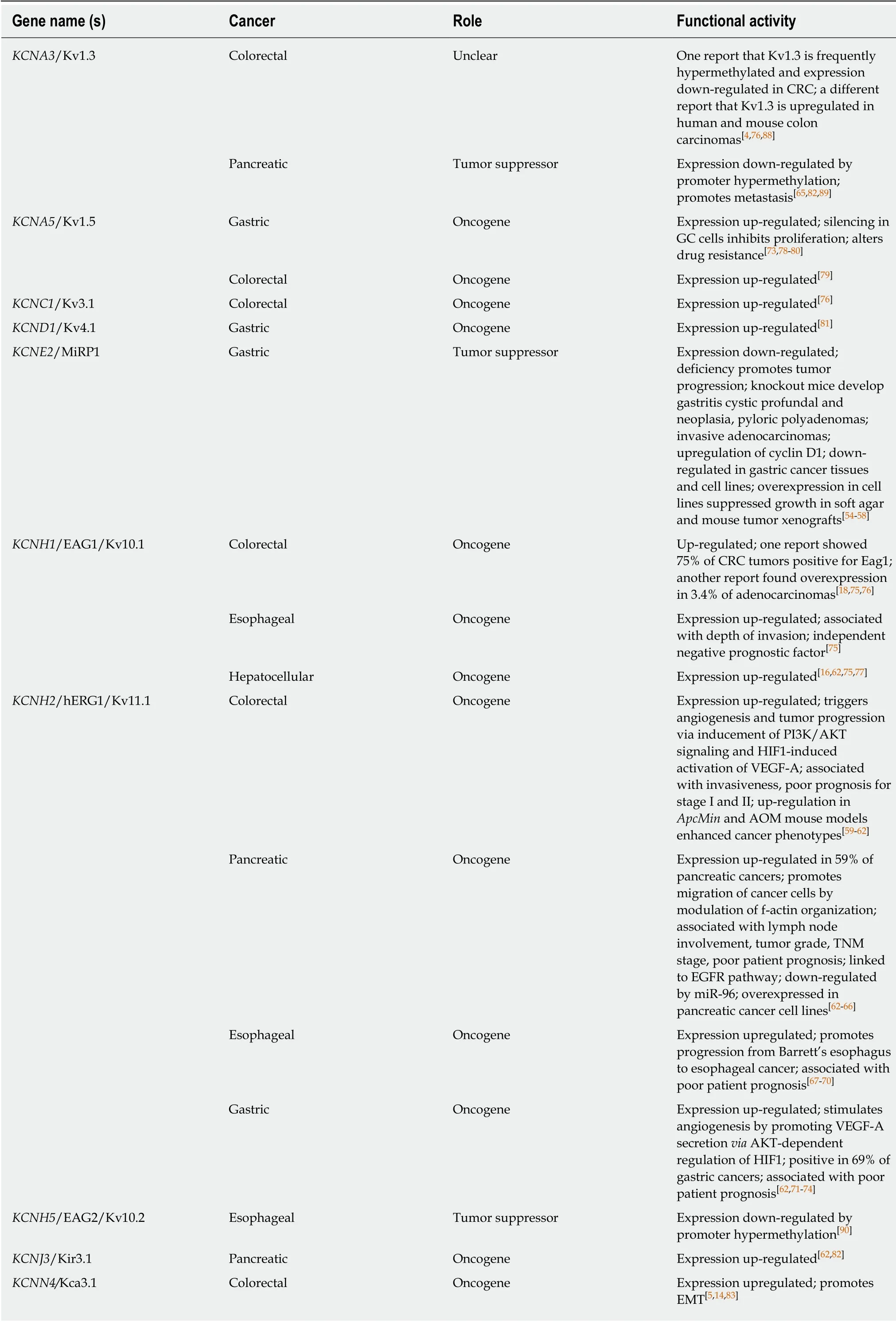
Table 1 Potassium channels
CRC: Colorectal cancer; GC: Gastric cancer; CIS: Common insertion site; EMT: Epithelial to mesenchymal transition; GI: Gastrointestinal.
CFTR is a tumor suppressor in CRC
The lifespan of individuals with CF has increased dramatically so that the average life expectancy of a person born with CF today is approximately 44 years[121]. As individuals with CF live longer it has become apparent that they are at increased risk for developing some but not all cancers. Initial evidence linking CF to cancer risk came from a 20-year epidemiological study that compared incidence of cancers in individuals in the United States Cystic Fibrosis registry to the predicted age adjusted risk in the general population to determine standardized incidence ratios. This study reported that the overall risk of cancer was not increased. However, the risk of all types of GI cancers was increased and in particular the risk of CRC, the most common GI cancer, was increased by 6-fold[122]. A recent meta-analysis of population-based studies is consistent with this result[123]. In support of the clinical significance of this finding, endoscopic screening studies of adult CF patients found that polyps in individuals with CF appeared earlier, and were larger and more aggressive than those in the non-CF population[124,125]. As a result of these studies the guidelines for endoscopic screening of CF patients have been modified and CF has been declared a hereditary colon cancer syndrome by the Cystic Fibrosis Foundation[126].
Mouse genetic studies
Mouse genetic studies demonstrated the functional significance of Cftr deficiency. SB transposon-mediated genetic screens initially identified Cftr as a candidate cancercausing gene. Cftr was identified in the top 10% to 50% of candidate genes in three SB screens to identify CRC driver genes in Apc wildtype[33], Apc-deficient[34], and TGF betadeficient[36]backgrounds[127]. Follow up studies evaluated mice carrying a targeted intestinal specific deletion of Cftr (Cftr-/-) for intestinal tumorigenesis. In the tumor sensitized ApcMinbackground Cftr-/-mice developed significantly more adenomas than ApcMinCftr wildtype mice. Further, ApcMinCftr-/-but not ApcMinCftr wildtype mice developed invasive lesions. Most striking, Cftr-deficiency alone was sufficient to cause adenomas in > 60% of mice after a one year interval[128].
CFTR deficiency in the general population
CFTR deficiency is linked to CRC in the general population as well. In a study of 90 Stage II CRC patients stratified by tumor CFTR expression, disease-free survival at 3 years in the 25% of patients with lowest CFTR expression was 30% lower than those with higher expression[128]. In a second cohort, CFTR mRNA and protein expression was lower in tumor vs normal tissue and CFTR mRNA expression was lower in metastatic vs non-metastatic tumors. In this study, CFTR-depleted CRC cell lines showed enhanced oncogenic characteristics including increased colony formation,migration and invasion[129]. RAS, PKC and IFNα have been reported to be involved in the down-regulation of CFTR in the colon[119]. It is also possible that low CFTR expression in sporadic tumors is caused by epigenetic silencing of the CFTR promoter as has been reported in other cancers such as lung and bladder[130-133].
CFTR and the stem cell compartment
The single cell layer of the intestinal epithelium is replaced approximately every 5 days. Stem cells at the base of the crypt drive this renewal[114]. These stem cells and progenitor cells that acquire stem-like characteristics are thought to be the CRC initiating cells[134]. In the colon, CFTR expression is highest at the base of the crypt[112]and expression has been reported in the stem cell itself[93]. CFTR has also been reported to be involved in intestinal lineage differentiation with its knockdown causing proliferation, migration and expression of EMT genes[135]. Thus, CFTR is in a position to influence the renewal process of the intestinal epithelium and CFTR deficiency may directly influence cancer initiating cells. Intestinal crypts can be cultured in vitro as 3D organoid cultures. Organoid cultures recapitulate the intestinal luminal epithelial structure as well as the renewal and differentiation process[136].CFTR is expressed in these cultures and maintains its ion channel function[137,138]. Thus,organoid cultures have been developed as surrogates to test the effectiveness of CF therapeutics as treatments for specific mutations[139-141]. In addition, organoid cultures derived from oncogenic or pre-oncogenic tissues maintain the oncogenic characteristics of these tissues[142,143]. Accordingly, organoid cultures have been used to determine the oncogenic characteristics of CFTR deficiency. Than et al[128]determined that Cftr-deficient colon organoids demonstrate increased clonogenicity. Analysis of small intestinal organoids by Strubberg et al[93]demonstrated increased proliferation of Cftr-deficient organoids and localization of Cftr to the LGR5 stem cell. These findings support a role for Cftr deficiency in the environment of the cancer initiating cell or the stem cell itself.
Wnt/β-catenin signaling
The Wnt/β-catenin signaling pathway is dysregulated in more than 85% of human CRC. Dysregulation contributes to initial adenoma formation and to maintenance of invasive tumors[144]. In the intestine CFTR deficiency is primarily associated with enhanced Wnt/β-catenin signaling. Than et al[128]identified nuclear localization of βcatenin, indicative of activation, as well as increased expression of Wnt/β-catenin target genes, in intestinal tumors deficient only for Cftr. Strubberg et al[93]identified increased active β-catenin in Cftr KO crypts and organoids which correlated with increased proliferation. Mechanistically, Cftr KO or transient inhibition of channel activity by CFTR(inh)-172 resulted in increased intracellular pH in LGR5+ stem cells.Increased pH in turn promoted association of Dvl2, a member of the Wnt/β-catenin signaling pathway, with PM phospholipids, which positioned it to enhance Wnt/βcatenin signaling[93](Figure 1A). In contrast, Chan and colleagues reported that intestinal Cftr deficiency in mice carrying the F508del mutation and knockdown of CFTR in the Caco-2 CRC cell line results in diminished β-catenin signaling.Mechanistically, loss of CFTR destabilizes β-catenin membrane localization allowing it to be degraded leading to oncogenic phenotypes via activation of NF-κB[145](Figure 1B). The differences between these two studies may reflect tissue specificity. This study analyzed Wnt/β-catenin signaling in total intestinal tissue rather than the crypt and so may reflect a different role for CFTR outside the stem cell compartment.Further, the effect of CFTR deficiency on Wnt/β-catenin activity varied by tissue in several other studies by this group[100]. In addition, Wnt/β-catenin signaling may also play different roles at different stages of CRC. Although it is generally accepted that dysregulated Wnt/β-catenin signaling is a driving force for initiation of CRC and progression[144]there is evidence that low levels of expression of Wnt/β-catenin targets are associated with poor prognosis in CRC[146]. Further, inhibition of Wnt/β-catenin has been shown to be necessary for survival of latent metastatic cells[147].
Additional mechanisms of tumor suppression
Extensive evidence that CFTR deficiency plays a role in many types of cancers suggests that tumor suppression by CFTR goes beyond regulation of Wnt/β-catenin signaling. The mechanisms of tumor suppression by CFTR are not well understood.However, the roles of CFTR in normal issue and the consequences of CF suggest plausible mechanisms.
Intestinal barrier integrity
CFTR plays a pervasive role in intestinal homeostasis through its influence on several inter-related processes: Epithelial barrier maintenance, microbiota composition and immune homeostasis. Breakdown of these processes is responsible for some of the clinical manifestations of CF and is also potentially oncogenic (Figure 2).
CFTR deficiency disrupts protective physical barriers
The intestinal lumen is home to trillions of bacteria. These bacteria provide essential nutrients and signals needed for intestinal epithelial and immune cell homeostasis.However, direct contact with the intestinal epithelium must be prevented. The colon epithelium is protected from direct contact by protective mucus layers and by tight junctions between the lateral surfaces of the single layer of epithelial cells lining the lumen. The apical, luminal surface of the epithelium is protected by a dense mucus layer impenetrable to bacteria that is generated by constitutive secretion of Muc2 from luminal goblet cells. This dense layer is partially enzymatically digested to generate a looser outer layer inhabited by commensal bacteria[148]. Muc2 is secreted in a condensed conformation that expands to form a fully functional mucus layer in the presence of extracellular HCO3-and H2O. CFTR ion channel activity is required for direct export of bicarbonate and also for indirect support of export of bicarbonate and water through other channels[149]. Loss of CFTR ion channel activity results in accumulation of dehydrated mucus in the distal small intestine and proximal colon,which causes intestinal obstruction seen in CF patients in the form of meconium ileus and distal intestinal obstruction[120]. In addition, severely dilated crypts are seen in the CFTR-deficient intestine due to accumulation of mucus in goblet cells which may reflect defects in secretion[150,151]. The CFTR-deficient mucus layer appears to allow dysregulated bacterial contact. Bacterial colonies are reported in crypts with accumulated mucus[150]. Further, comparison of gene expression in the Cftr-deficient mouse intestine to changes in Muc2-deficient intestine using gene set enrichment analysis (GSEA) showed enrichment in inflammatory gene expression changes[128]suggesting that Cftr deficiency, like Muc2 deficiency, allows illicit bacterial contact.The basolateral epithelial surface and underlying lamina propria, including resident immune cells, are protected from bacterial contact by tight junctions and CFTR participates in maintenance of these junctions. In Cftr-deficient mice the small intestine displays increased intestinal permeability and disruption of tight junctions[152]. Several studies link CFTR interactions mediated by its C-terminal PDZbinding domain to integrity of tight junctions[152]. Studies in cultured airway epithelial cells show that interaction between the CFTR-PDZ binding (CFTR PDZBD) domain and the PDZ domain of NHERF1 (SLC9A3R1) maintains actin cytoskeletal organization and tight junctions[153]. CFTR deficiency may also maintain tight junctions by via direct interaction with ZO-1 an essential component of these junctions[8].
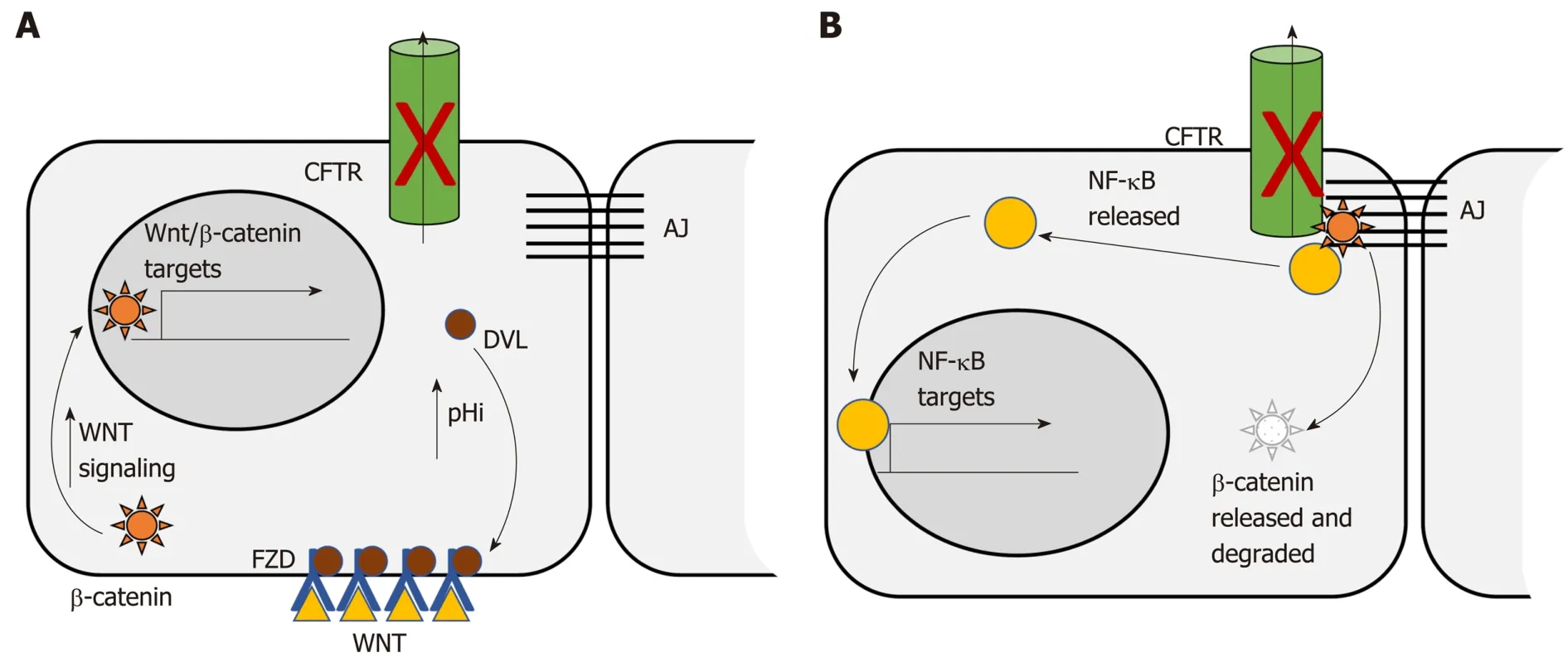
Figure 1 Two models for the effect of CFTR deficiency on Wnt/β-catenin signaling. A: CFTR deficiency promotes Wnt/β-catenin signaling. CFTR deficiency causes increased intracellular pH. Increased pH promotes association with Dishevelled (DVL) at the membrane and with the Wnt receptor Frizzled (FZD). DVL association with FZD enhances Wnt/β-catenin signaling leading to increased nuclear localization of β-catenin. Nuclear β-catenin promotes transcription of genes involved in proliferation, survival and stemness[93]; B: CFTR deficiency inhibits Wnt/β-catenin signaling. CFTR deficiency releases membrane associated β-catenin to the cytosol where it is degraded thus decreasing Wnt/β-catenin activity. Loss of β-catenin releases NF-κB which translocates to the nucleus where it promotes transcription of inflammatory targets[145]. FZD: Frizzled; DVL: Dishevelled; AJ: Adherens junctions.
CFTR deficiency causes dysbiosis
As described above, CFTR deficiency as seen in CF patients and animal models creates an altered luminal environment. This environment, as well as a high fat/high calorie diet maintained to overcome nutritional deficits, and CF therapies such as frequent antibiotic exposure, contributes to bacterial dysbiosis[154,155]. In early studies dysbiosis was reported as small intestinal bacterial overgrowth in a CF mouse model[156]. Analysis of microbial 16S DNA in small intestine flushed luminal contents by qRT-PCR demonstrated an estimated 40X increase in bacterial load with decreased diversity[157]. In a second study using phylogenetic microarray analysis of flushed small intestinal contents there was a marked decrease in bacterial community richness, evenness and diversity, a decrease in the relative abundance of protective species such as Acinetobacter Iwoffii and Lactobacilliales members, but an increase in Mycobacteria species and Bacteriodes fragilis associated with GI infection and immunomodulation[156]. More recently, microbial population composition of CF vs non-CF individuals has been determined using targeted and metagenomic analysis of 16s rRNA DNA from fecal DNA[94,95,155,158]. Further, CF patients have been reported to be susceptible to Crohn's Disease with specific CFTR mutations influencing microbial dysbiosis with increased intestinal permeability[159]. In each of these studies CF microbiota demonstrated greatly reduced diversity and significant differences from non-CF controls indicating that the altered luminal environment not only creates increased bacterial access but also changes bacterial composition[95,103,156]. Importantly,alterations to the intestinal microbiome are increasingly associated with CRC[103,160].
CFTR deficiency is associated with immune infiltration
Disruption of the mucus layers and of tight junctions allows bacterial access to immune cells and immune infiltration of the epithelial layer. Although immune infiltration and subsequent inflammation can cause overt damage, this effect has only been reported in one study in which a capsule endoscopy study of CF patients revealed duodenal lesions[161]. More commonly, immune infiltration has been detected under CFTR-deficient conditions but has not been accompanied by obvious morphological and histological damage[162]. In human studies CF patients exhibited evidence of immune infiltration in studies evaluating whole gut lavage[163], fecal calprotectin[158,161,164]and rectal nitric oxide[165]. Further, microarray analysis of the small intestine in an early study by Norkina et al[162]identified upregulation of genes expressed by granulocytes which was supported by microscopy that identified increased mast cells and neutrophils throughout the length of the intestine. Similarly,Chan and colleagues reported immunocytochemical evidence of an increased presence of macrophage and neutrophils in the small intestine of F508del mice[145].
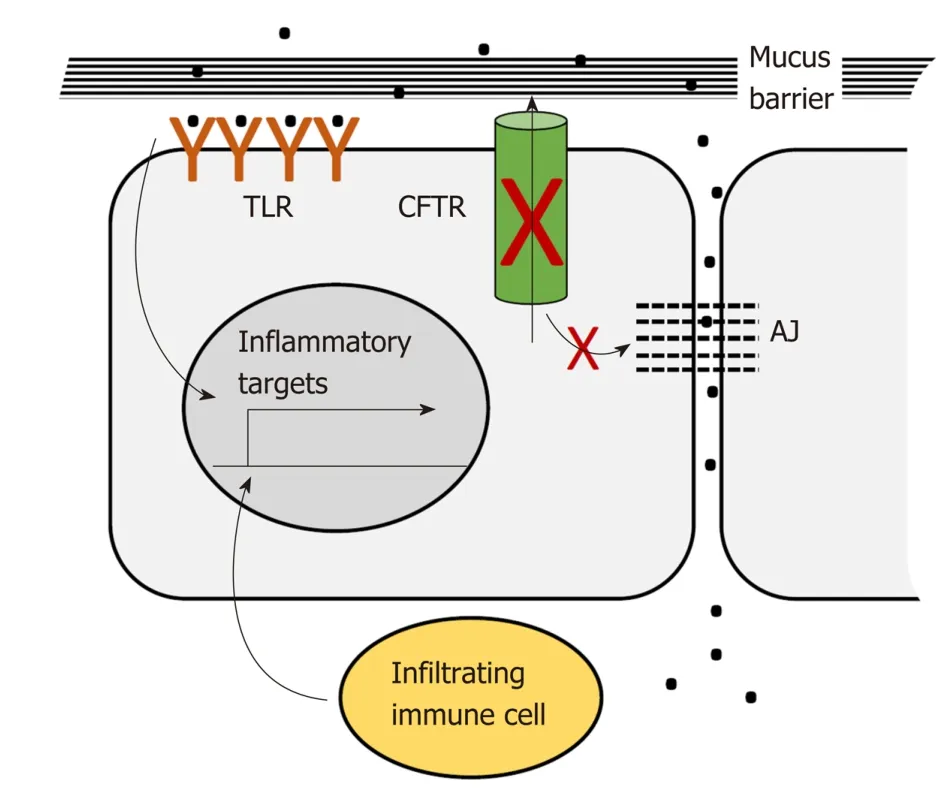
Figure 2 CFTR deficiency disrupts epithelial barrier integrity. CFTR deficiency disrupts the mucin barrier and adherens junctions. This allows bacterial contact with the apical and basal surfaces of the epithelial layer. Contact with the apical layer stimulates inflammatory signaling via toll-like receptors. Contact with the basal layer leads to immune cell infiltration which results in additional inflammatory signaling. AJ: Adherens junctions; TLR: Toll-like receptors.
Potential oncogenic changes
Loss of physical barriers in the GI tract allows dysregulated access of bacteria and infiltration of immune cells both of which contribute to inflammatory signaling[166-169].These contacts have the potential to activate pro-inflammatory signaling in the intestinal epithelium both directly by interaction between bacteria and epithelial tolllike receptors[170]and indirectly via activation of lymphocyte cytokine signaling. For example, Fiorotto et al[171]demonstrated that in primary biliary epithelial cells CFTR binds Src inhibitors to position them for interaction with Src. Loss of CFTR leads to mislocalization of these inhibitors and activation of Src. In the presence of bacterial products this leads to NF-κB signaling which ultimately disrupts tight and adherens junctions. Proinflammatory signaling by intestinal epithelial cells is associated with dysregulated proliferation and expansion of the stem cell compartment through reversion of progenitor cells to stem cells[172]. Loss of barrier integrity affects other processes as well. Loss of tight junctions facilitates increased migration and invasion[173]. Dysbiosis may result in appearance of bacterial species associated with CRC. Phylogenetic microarray analysis of Cftr-deficient mice detected increased representation of Bacteroides fragilis[156], a species directly linked to CRC by virtue of its activation of Stat3 signaling through a Th17 immune response[174].
In vitro models of pro-inflammatory signaling
CFTR deficiency has been linked to NF-κB activation in in vitro models. Chan and colleagues have carried out a series of studies in cell lines derived from several cancers, that delineate a pathway in which CFTR deficiency leads to activation of NFκB, transcription of UPA, and enhanced migration and invasion[175-177]. Other groups have shown increased basal levels of pro-inflammatory cytokines and other factors and NF-κB pathway members in CFTR-deficient Caco-2, HT-29 and HEK293 cells and increased response to inflammatory stimuli, including an increase in TNF-α, IL-6, IL-1β-induced secretion of IL-8, COX-2 and PGE2,plus increased activities of ERK1/2,MAPK, IκBα and NF-κB[92,96,178]. Santa-Coloma and colleagues report activation of NFκB as a result of autocrine signaling by IL-1β[179]. In addition, the Hedgehog pathway has recently been reported to be inhibited under CFTR-deficient conditions which is predicted to activate inflammatory signaling[180].
Stress responses
CFTR deficiency has been reported to both increase oxidative stress via impaired mitochondrial activity[96,97]and to reduce stress by cellular retention of the antioxidant GSH[181]. In Caco-2/15 cells CFTR knockdown caused an increase in lipid peroxidation levels accompanied by a decrease in oxidant defenses such as glutathione peroxidase and catalase[182]. In addition, loss of CFTR has been reported to disable autophagy via activation of Transglutaminase-2 whose cross-linking activity causes essential autophagy proteins to be sequestered in aggresomes[183]. Finally, HIF-1-mediated repression of CFTR in intestinal epithelium resulted in a reduction in CFTR mRNA,protein and activity in hypoxic epithelium[184]. The impact of these activities on oncogenesis is not yet known.
Relationship to CRC
CFTR deficiency results in dysregulation of many processes that may be oncogenic.However, the relative contributions of these processes to the development of CRC in vivo are not yet clear. In the future it will be important to study CFTR in the environment in which it most likely contributes to development of CRC, the intestinal stem cell compartment. In the colon, CFTR is most highly expressed in the base of the crypt which comprises the stem cell compartment and the likely site of origin of CRC.Because the crypt is the environment for the intestinal stem cell it has unique protective mechanisms. The crypt maintains a unique microbiota with reduced number and diversity compared to the lumen[185]. Limited contact between bacterial products and crypt epithelial cells contributes to this protected environment. For example, goblet cells at the neck of the crypt orchestrate the release of mucin in response to contact with bacterial products[186]. The crypt also has unique mechanisms to protect from cellular stress. In the stem cell, interaction between the cytosolic innate immune sensor NOD2 and the bacterial peptidoglycan motif MMP is necessary for stem cell survival in the face of oxidative stress[187]. Finally, cells in this compartment are uniquely susceptible to the effects of inflammatory signaling. In a mouse genetic study NF-κB was shown to synergize with Wnt/β-catenin in intestinal crypt cells to promote conversion of progenitor cells to stem-like cells with tumor initiating capacity[172]. These considerations highlight the importance of studying potential oncogenic phenotypes of CFTR deficiency in the crypt and crypt-derived organoid models.
CFTR and other cancers
Given the widespread distribution of CFTR and its impact on cellular homeostasis it is not surprising that dysregulation of CFTR is implicated in many cancers. CFTR overexpression has been correlated with cancer in individual studies, in particular,gastric and ovarian cancers[101,188-192]. However, in the vast majority of studies CFTR deficiency is associated with cancer occurrence, and in most of these studies, cancers with CFTR mutations or down-regulation of expression in tumors are more likely to exhibit rapid expansion, EMT, decreased apoptosis, increased metabolic potential,and increased patient morbidity and mortality. Decreased CFTR expression and/or inactivating mutations are associated with non-small cell lung cancer[130,175,193],glioblastoma[194], bladder[131-133], EC[195,196], PC[197,198], nasopharyngeal[199], prostate[176], and breast[177]cancers. In most of these cancers CFTR deficiency was associated with increased tumor stage and poor survival. Germline CFTR mutations, as seen in CF,have been linked to increased risk of PC among younger patients[197,198]. Germline mutations may be an important risk factor in the general population as an estimated 3% of the United States population are heterozygote carriers for deleterious CFcausing CFTR mutations[200]and therefore potentially at risk due to loss of heterozygosity or haploinsufficiency. However, in most studies CFTR deficiency was associated with differentially decreased expression but not specifically with germline defects. Although germline mutations were not examined in these cases it is unlikely that decreased expression was caused primarily by CF-causing germline mutations because F508del, which makes up approximately 70% of CF alleles, causes a 90%decrease in protein levels but only a modest decrease in mRNA. Known causes of decreased CFTR expression include hypermethylation as seen in lung and bladder cancer studies[130-133]and somatic mutations seen in a NSCLC study[130]. In addition,cigarette smoke (CS) has recently been shown to down-regulate CFTR expression and CFTR down-regulation has been linked to the etiology of COPD[201]. However, a recent study also linked CS-mediated down-regulation of CFTR to exacerbation of oncogenic phenotypes in the A549 lung cancer cell line[202].
CFTR therapies and translational and clinical cancer implications
In the era of precision medicine new CF modulator therapies targeted to specific CFTR mutations have entered the clinic. These include potentiator drugs that increase anion flow through CFTR channels already present on the PM and corrector drugs that promote correct folding of mutant proteins. Three modulators, ivacaftor,tezacaftor, and lumacaftor, have been approved for treatment of CF[203]. Ivacaftor is a potentiator approved for treatment of > 25 CF-causing mutations including gating,residual function and splice mutations. Tezacaftor and lumacaftor are correctors designed to improve the function of the F508delta mutation which makes up approximately 70% of CF alleles. As discussed by Bodewes et al[204]these drugs are finally targeting CF-related GI diseases, with potential use in cancer therapy. Some examples include treatment of pancreatitis with Ivacaftor[204], drug treatment of CF patients that improved proximal small intestine pH as a regulator of bicarbonate secretion, improvement in cell motility and clinical outcomes in patients with CFTR G551D mutations[104], improvement in bicarbonate permeability following Lumacaftorrescued F508del mutations[205], improvement in gut microbiota and intestinal inflammation following treatment with Ivacaftor, including an increase in Akkermansia and a decrease in Enterobacteriaceae, and a significant reduction in inflammation in patients treated with Ivacaftor[206]. Additional modulators and combinations of modulators are under development such as the combination of Ivacaftor with 5-Nitro-2-(3-Phenylpropylamino) Benzoate, that is reported to act synergistically in activation of G551D mutant CFTR[207]. These mutation-specific drugs are potentially applicable to the 3% of Caucasians who are CF carriers, having one CF-causing CFTR mutation.These individuals are potentially at risk for developing CRC due to haploinsufficiency or loss of heterozygosity. In addition to these Food and Drug Administration (FDA)-approved therapies, another category of modulator known as amplifiers, represented by PTI-428, is in clinical trials. This drug is designed to enhance translation of CFTR mRNA to increase protein production and facilitate the activity of corrector drugs[99].Thus, this drug has the potential to improve CFTR activity in sporadic CFTR-low CRC. In addition, testing strategies used for screening and diagnosis of CF may be applicable to early detection and even prevention of CRC. Currently genetic testing for CF-causing mutations is recommended for all pregnant couples. If carrier status proves to be a risk factor for CRC, then this genetic testing may be adaptable for early detection of CRC as well. A significant technical advance to test new CF therapies has been the development of patient-derived colorectal organoids. An example is the work of Dekkers et al[208]who have demonstrated proof of this idea in rectal organoids derived from CF patients to test a range of CF drugs, including Ivacaftor and Lumacaftor. While these new targeted CF therapies have yet to enter the cancer clinic,they may soon do so, along with the repurposing of other ion channel activators and blockers as more becomes known about the precise contribution of ion channels to cancer development.
Ca2+ activated Cl- channels
Ca2+activated Cl-channels (CLCAs) are a family of secreted self-cleaving proteins that activate Ca2+-dependent Cl-channels. CLCAs are involved in Cl-conductance across epithelial cells and therefore influence epithelial fluid secretion, cell-cell adhesion,apoptosis, cell cycle control, tumorigenesis and metastasis, mucus production and cell signaling[209,210]. There are four CLCAs (1-4) in humans and two of these family members (CLCA1,2) are implicated in GI cancers, almost invariably as tumor suppressors whose down-regulation is associated with cancer progression and poor patient prognosis. CLCA1 is down-regulated in CRC and this is associated with poor patient prognosis[4,211,212]. CLCA1 has also been reported to suppress CRC aggressiveness via inhibition of the Wnt/β-catenin signaling pathway[213]. CLCA1 is also reported to be a prognostic biomarker for PC, with lower CLCA1 expression correlated with shorter disease-free survival[214]. CLCA2 is also down-regulated in CRC[4,212].
Cl- intracellular channels
Cl-intracellular channels (CLICs) are novel, auto-inserting, self-assembling intracellular anion channels involved in a wide variety of fundamental cellular events including regulated secretion, cell adhesion, cell cycle and apoptosis. However, the actions of CLICs remain to be fully explained. They are a class of intracellular anion channels that do not resemble classical ion channel proteins. CLICs can exist as both soluble globular proteins and integral membrane proteins with ion channel function.Human CLIC1 adopts at least two stable soluble structures, with redox status controlling the transition between them. CLIC proteins are characterized by the presence of a 240 residue CLIC module whose structure belongs to the glutathione Stransferase fold superfamily[215]. Three CLICs appear to be functional in humans(CLIC1,3,4) with two CLIC proteins (CLIC1 and CLIC4) that appear to be essential molecular components of anion channels, with CLIC1 capable of forming anion channels in planar lipid bilayers in the absence of other cellular proteins. However,these putative ion channel proteins are controversial because they exist in both soluble and membrane forms, with at least one transmembrane domain[209,215]. All three CLICs are involved in GI cancers, with all implicated as oncogenes. CLIC1 is overexpressed in CRC[216-219]; in PC[65,217], where it was reported to be upregulated in 69% of tumors and associated with poor patient prognosis; and in GC[65,218,220-223], where it was found upregulated in 68% of tumors, correlated with lymph node metastasis,lymphatic invasion, perineural invasion and poor patient prognosis. It was also reported to promote GC progression by regulating ROS-mediated MAPK/AKT signaling. CLIC1 was also reported to be upregulated in HCC[65,224]and gall bladder cancer[225,226]. CLIC3 is upregulated in PC where it was reported to promote integrin recycling from late endosomes to drive PC progression[65,227]. CLIC3 is also a secreted protein that is reported to drive cancer progression through its glutathione-dependent oxireductase activity. In particular CLIC3 was identified as part of the cancer associated-fibroblast secretome where it promotes the invasive behavior of endothelial cells to promote angiogenesis and invasion. CLIC3 is also secreted by cancer cells[228], and CLIC3 is described as a pH sensor, important as changes in cellular pH influence cell proliferation and the balance between cell survival and cell death[229]. CLIC4 is upregulated in CRC where it was found to be a direct response gene for C-MYC and TP53, with its overexpression associated with poor 5-year patient survival[218,230]. CLIC4 is also upregulated in PC with its expression associated with tumor grade, invasion and poor patient survival[231]. CLIC4 was also found to be expressed in mitochondria where it regulates pH and cell volume.
ANO1
ANO1 is also referred to as Anoctamin-1, DOG1 and TMEM16A. ANO1 is a Ca2+-activated Cl-channel that mediates receptor-activated Cl-currents that are involved in a range of physiological processes. ANO1 is activated by intracellular Ca2+. ANO1 has 8 putative transmembrane domains and demonstrates no similarity to other ion channels. It is expressed in a variety of secretory epithelia, including the gut. ANO1 activity is implicated in regulating CFTR Cl-channel activity. In all of the GI cancers studied, ANO1 expression and activity has been reported to upregulated, thus ANO1 can be described as an oncogene in the GI tract. In CRC, ANO1 expression is upregulated, associated with EMT and poor patient prognosis[232-236]. In PC, ANO1 expression is important for cell migration[234,237]. In EC, ANO1 is a biomarker of EC progression[234,238]. In GC, its expression is upregulated[234,239], and in GI stromal tumors,ANO1 expression is used as a diagnostic biomarker, and it is associated with negative regulation of IGFBP5[240-242]. See Table 2 for a full listing of chloride ion channels in GI cancers.
CALCIUM CHANNELS
Ca+is a ubiquitous second messenger, serving as a signaling molecule for a variety of cellular processes such as control of the cell cycle, apoptosis, and migration. Ca+concentrations are tightly regulated within the cell, with basal cytoplasmic levels being many orders of magnitude less than in the extracellular space. This control is essential for cellular homeostasis[253]. In addition to extracellular Ca2+, the endoplasmic reticulum (ER) and mitochondria are also significant stores of Ca2+. Selective distribution and activation of Ca2+channels at any of these sources allows for Ca2+micro-domains and adds another level of specificity to Ca2+signaling[6]. Because it is involved in such a broad array of processes, and signaling molecules are often sensitive to very minute changes in Ca2+, it is easy to see how perturbations in Ca2+handling could lead to significant physiological outcomes.
Ca2+signaling is typically initiated through the ligand activation of various receptors that activate phospholipase C[254]. This leads to the production of inositol triphosphate, which diffuses to the ER, where it binds to and opens its receptor and Ca2+channel. Upon its release into the intracellular space, Ca2+binds to calmodulin and a variety of other Ca2+-activated proteins to elicit a wide variety of responses.Over time, these intracellular stores would be depleted if not for being replenished by extracellular Ca2+. The release and depletion of ER Ca2+triggers the process of storeoperated calcium entry (SOCE), in which PM Ca2+channels are opened in order to allow entry of extracellular Ca2+[254]. One of the proteins responsible for sensing the depletion of ER stores, and initiating SOCE, is stromal interaction protein 1(STIM1)[254]. In CRC, STIM1 over-expression has been associated with increased tumor size, depth of invasion, lymph node metastasis, and increased serum levels of carcinoembryonic antigen[15,255,256]. When injected into immunocompromised mice,CRC cells expressing a higher level of STIM1 had a much higher incidence of lung and liver metastasis than those expressing lower levels[257].
The PM Ca2+channels opened by STIM1 activation include ORAI1 as well asmembers of the TRP superfamily of cation channels. In addition to replenishing intracellular stores, these PM channels also contribute to Ca2+signaling in response to intracellular or extracellular cues. Different members of the TRP superfamily are activated by physical changes such as mechanical stress, osmotic pressure, or temperature; or chemical changes such as pH, growth factors/cytokines, pO2, or ROS[258]. With all of these being central factors in a tumor microenvironment, how a cell alters its response to these conditions could ultimately influence the death or survival of a potentially cancerous cell.
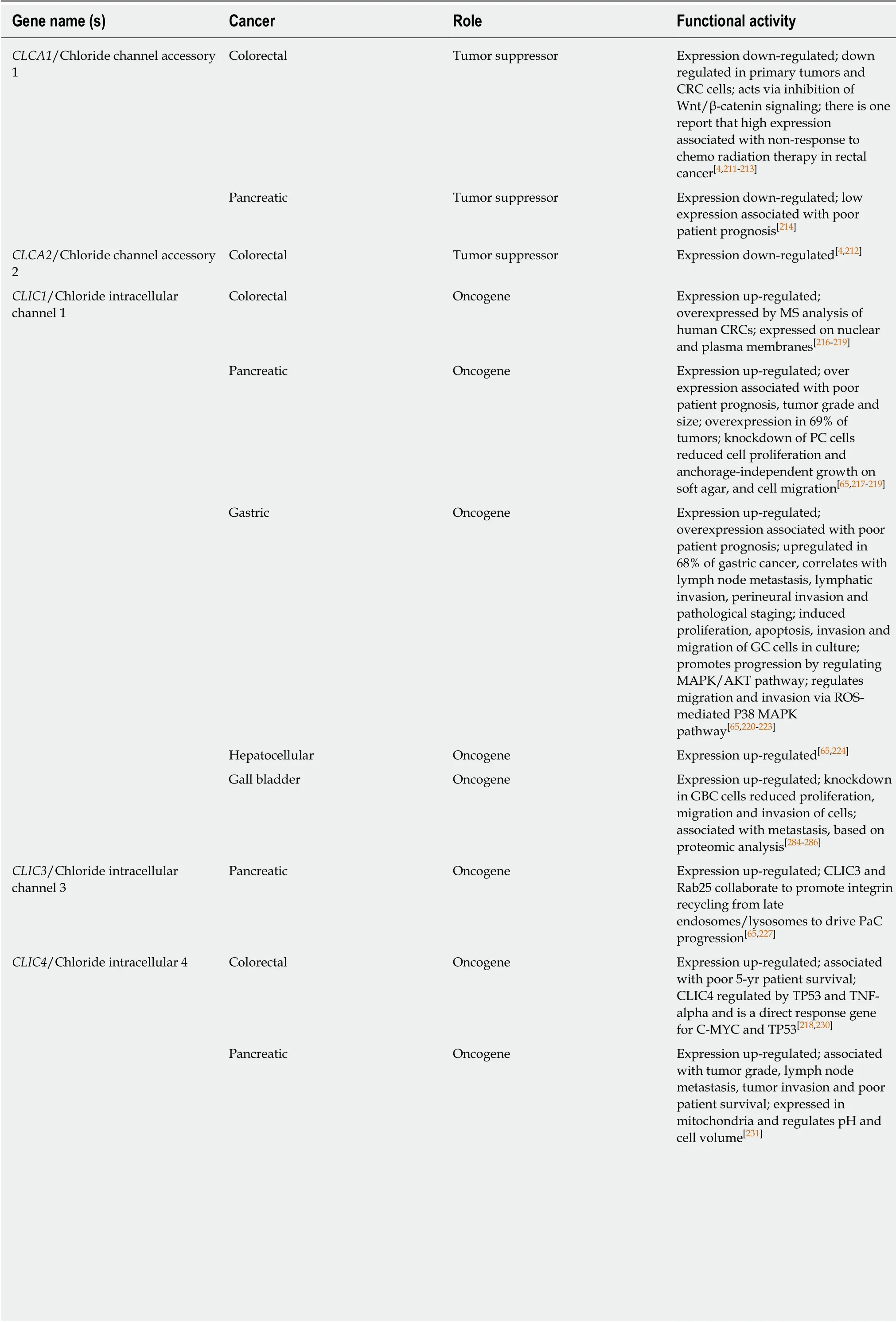
Table 2 Chloride channels

CRC: Colorectal cancer; PC: Pancreatic cancer; GC: Gastric cancer; GBC: Gall bladder cancer; EMT: Epithelial to mesenchymal transition; EC: Esophageal cancer; GIST: Gastrointestinal stromal tumors; CF: Cystic fibrosis.
Transient receptor potential family
Various members of the transient receptor potential (TRP) superfamily have been implicated in GI cancers, in particular the TRPM (m = melastatin) and TRPC (c =canonical) sub-families. The cold-activated TRPM8, well known for its role in androgen-dependent prostate carcinoma, is also over-expressed and necessary for the proliferation and migration of PC cells[259-262]. TRPM7 also plays a role in PC, but by increasing motility and thus the potential for metastasis, with its depletion causing a decrease in invasiveness through the HSP90α/uPA/MMP-2 proteolytic axis and targeted senescence, while results vary as to its role in proliferation[5,258-261,263-267]. TRPM7 has also been implicated as an oncogene in CRC[268]and GC[269-271]. Though a specific mechanism has not been proposed, TRPM2 over-expression has also been shown to reduce PC as well as GC patient survival by increasing invasiveness and proliferation[272,273]. Expression of TRPC1 is reported to be upregulated in CRC[5,256,274],where it promotes metastasis, EC[275], and GC[68]. TRPC6 expression is upregulated in EC[276,277]where it was found to be necessary for Ca2+increase to promote G2 progression, and association with tumor stage and poor patient prognosis, GC[5,278],and HCC[4].
ORAI1
Through its contribution to hyperactivity of intracellular Ca2+oscillations, ORAI1-high cells had increased activation of ERK, AKT, and myocyte enhancer factor 2D,indicating a possible mechanism explaining their increased proliferative and invasive phenotype[275]. ORAI1 expression is also upregulated in CRC[5], where it is activated by STIM1; PC[92,279], where it mediates SOCE and promotes apoptotic resistance in PC cells; GC[68], where it is associated with promoting metastasis; and in EC, where ORAI1 over-expression had a negative effect on patient prognosis.
Other oncogenic Ca2+channels include SIGMAR1 in CRC[280-282], L-TYPE CACNA1C in CRC[283], T-type CACNA1I in GC[284], where its upregulation is associated with poor patient survival, and T-type CACNA1H in GC[284], where it is also associated with poor patient survival.
Tumor suppressor Ca2+ channels
While most Ca2+channels appear to function in an oncogenic fashion when dysregulated there are several exceptions. TRPM6 has been reported to be downregulated in CRC (16 of 20 tumors studied) with high expression associated with better patient survival[285]. STIM2 is reported to be down--regulated in CRC leading to apoptosis resistance[15,256,274]. Expression of the T-type channel CACNA1G is reported to be down-regulated by promoter hypermethylation in CRC[286], PAC[287], and GC[288],with high expression associated with improved overall survival. CACNA2D3 expression is down-regulated by promoter hypermethylation, associated with worse patient prognosis[91,289]and expression of CACNB2 is also reported to be downregulated by promoter hypermethylation[90]. See Table 3 for a full listing of the role of calcium ion channels in GI cancers.
SODIUM CHANNELS
Voltage-gated sodium channels (VGSCs) are classically associated with the initiation and conduction of action potentials in electrically excitable cells such as neurons and muscle cells. They are typically heteromeric complexes composed of one of 9 poreforming α-subunits and one of 5 regulatory β-subunits, though the α-subunit can usually be a functional channel by itself. These channels have recently been discovered in non-excitable cell types such as glia, fibroblasts, immune cells, and cancer cells, where their function is less understood[290]. In the developing central nervous system, VGSCs regulate the migration and pathfinding of neurite outgrowth[291-293]. Similarly, in the vast majority of cancers where these channels have been studied, their major influence has been to increase the motility and invasiveness of cancer cells.
The VGSC NaV1.5 is abundantly expressed in human CRC cells SW620, SW480, and HT29, as well as being highly expressed in tumor biopsies relative to adjacent normal tissue[294]. Through pharmacological inhibition, siRNA knockdown, and controlling for Ca2+influence, House et al[294]demonstrated that conductance through NaV1.5 contributes significantly to CRC cell invasiveness and cancer progression. This group later demonstrated this contribution mechanistically through correlating NaV1.5 activity to RAP-1 mediated MAPK activation and changes in gene expression through the PKA/ERK/c-Jun/ELK-1/ETS-1 signaling pathway[295]. While this group demonstrated the metastatic contribution of NaV1.5, Peng et al[296]have recently shown Nav1.5 expression to be predictive of clinical outcomes in non-metastatic CRC, with the potential involvement of estrogen receptor (ER)-β, implicating multiple roles for the same channel in different stages of cancer progression.
In GC, high expression of the VGSC Nav1.7 has similarly been shown to correlate with higher recurrence and reduced survival[297]. The detailed mechanism proposed by Xia et al[297]involves increased expression of the oncoprotein metastasis-associated incolon cancer-1 (MACC1) by Nav1.7 activation in a p38/NFkB-dependent manner.Increased MACC1 expression subsequently leads to HGF/c-Met/c-Jun-dependent activation of another oncoprotein, the Na+/H+exchanger-1 (NHE1) which, through its involvement in maintaining intracellular and extracellular pH, has been shown elevated in a variety of cancers[298-301]. Reports involving other Navchannels in GI cancer are fewer than for Nav1.5 and Nav1.7. Nav1.1 has been reported to be upregulated in CRC[302], associated with a shorter time to cancer recurrence in stage II and III disease. In contrast, there is one report of Nav1.6 being down-regulated in CRC[303].
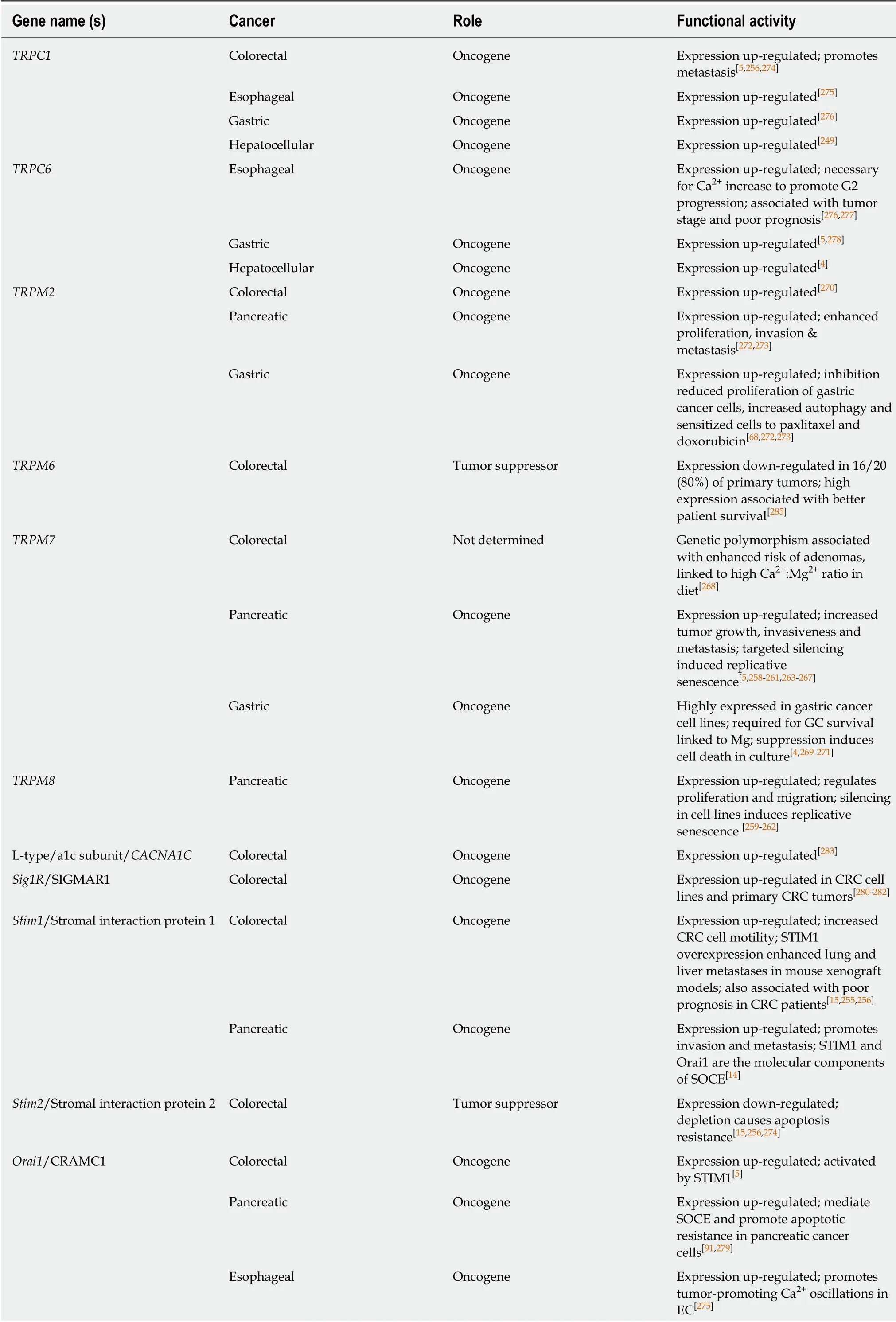
Table 3 Calcium channels

GC: Gastric cancer; CRC: Colorectal cancer; EC: Esophageal cancer; SOCE: Store-operated calcium entry; STIM1: Stromal interaction protein 1.
The mechanisms through which VGSC α and β subunits contribute to cancer progression typically differs according to their physiological functions: α subunits,through the conduction of Na+currents, and β subunits through the regulation of adhesion interactions, though contributions of any β subunit have yet to be shown in a GI cancer. The reason that only α subunits have been associated with GI cancers may owe in part to these differences in function relative to the demands of GI epithelial physiology. This specificity may also prove advantageous in pharmacologically targeting these subsets of cancers, as drugs targeting α subunit function have proven effective in reducing metastasis in murine xenograft breast cancer models[304,305]. See Table 4 for a full listing of sodium ion channels in GI cancer.
ZINC TRANSPORTERS
Zinc is a key trace element in the human body. Approximately 10% of human genes have the potential for zinc binding and a large number of human proteins (as many as 3000), including transcription factors, a variety of receptors, kinases, ligases and other enzymes, require zinc for their catalytic activity or tertiary structure[306,307]. Zinc exists in cells as Zn2+ions that are maintained in this valence state under all biologically relevant redox potentials and pH conditions in a cell. Zinc mediates a wide range of cellular processes that are important for maintenance of tissue homeostasis[306,308,309].Therefore, alterations in cellular zinc levels, such as zinc deficiency, can disrupt cellular function[306,308]. Furthermore, zinc is toxic to cells, thus zinc levels are tightly regulated within cells. As zinc cannot freely pass across cellular membranes a variety of Zn2+-permeable channels and transporters regulate Zn2+entry and exit from cells[306].Zinc transporters include both influx and efflux transporters[306,310,311]. Zinc also enters cells via other ion channels, such as Zn2+activation of several of the Zn2+-permeable voltage-gated Ca2+ion channels[309]. Examples include the store-operated Ca2+entry(SOCE) channels and the TRP family of Ca2+channels (discussed earlier in this review).
Here, the review will focus on the functions of the two major families of Zn2+transporters: ZnT/SLC30A and ZIP/SLC39A. Members of both of these families of proteins are reported to be dysregulated in human cancers, including GI cancers.There are 14 ZIP proteins (ZIP1-ZIP14) and 10 ZnT proteins (ZnT1-ZnT10) in thehuman body with differential tissue-specific expression[310,311].
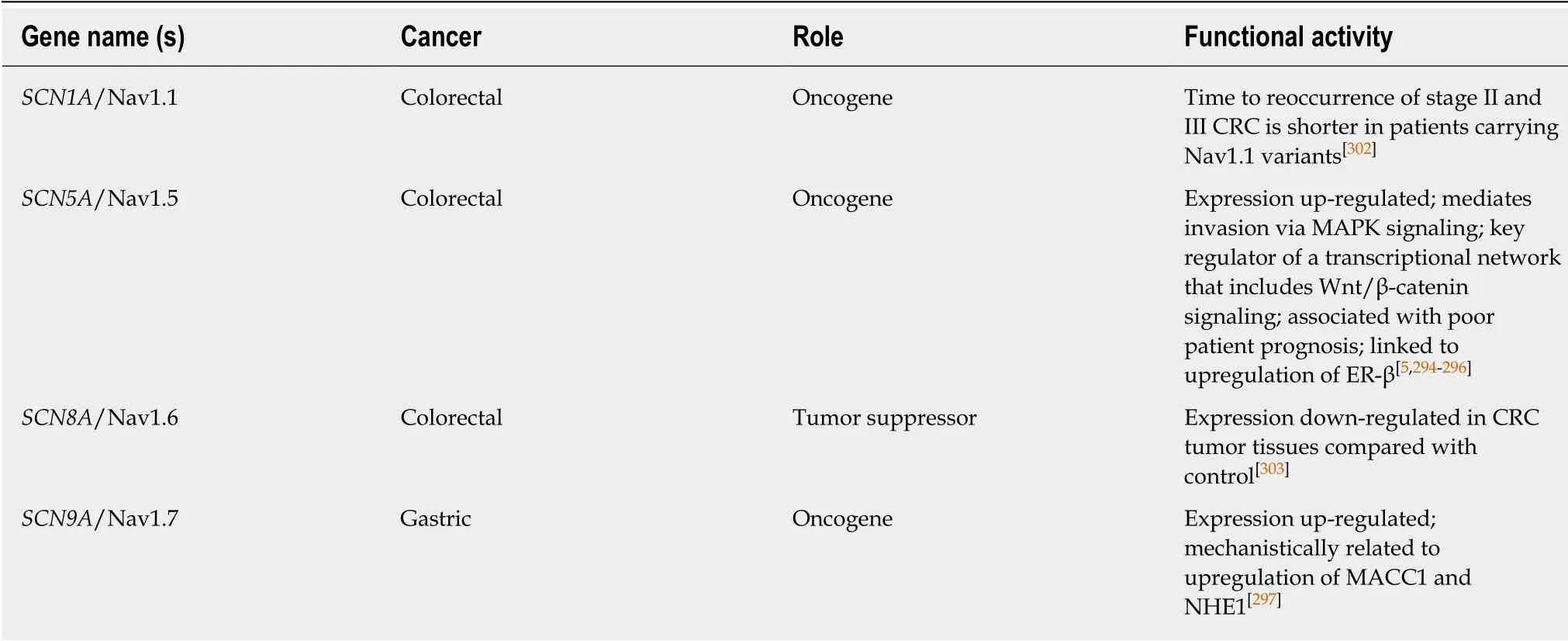
Table 4 Sodium channels
ZIPs
These proteins are encoded by the solute carrier family 39A genes (SLC39A1-SLC39A14). ZIPs mainly participate in the uptake of Zn into the cytoplasm from the extracellular space or from intracellular compartments such as the ER, Golgi or mitochondria. In the GI tract ZIPs 1, 3, 4, 5, 6, 7, 8, 9, 10, 11 and 14 are reported to be normally expressed in the intestines; ZIPs 1, 3, 4, 5, 6, 7, 8, 9, 10, and 14 in the pancreas; ZIPs 1, 2, 3, 5, 6, 7, 8, 9, and 14 in the liver; and ZIPs 1, 3, 4, 5, 6, 7, 8, 9 10,and 11 in stomach[310,312,313].
ZnTs
These proteins are encoded by the solute carrier family 30A genes (SLC30A1-SLC30A10). ZnTs participate in the efflux of Zn from the cytoplasm into the extracellular space or to sequester Zn in intracellular compartments. In the GI tract,ZnT1 expression is normally expressed ubiquitously; ZnTs 2, 3, 5, 6, 7 and 10 are normally expressed in the intestines; ZnTs 2, 3, 5, 6, 7 and 8 in pancreas; and ZnT 5, 6,7 and 10 in liver and ZnT 6, and 7 in stomach[310,312,313].
Zinc and zinc transporters in the GI tract
It is well known that dysregulation of zinc homeostasis results in GI dysfunction[311,312,314,315]. Examples include zinc deficiency resulting in diarrhea[311], and inflammatory bowel disease[316], clinical problems that are ameliorated by zinc supplementation[311,316,317]. This effect of zinc supplementation may be mediated by a zinc sensing receptor molecule, GPR39, that is similarly shown to be protective in the colon[318]. Zinc is also reported to enhance tight junctions and intestinal mucosal barrier functions in general[319,320]. Maintenance of zinc homeostasis is primarily mediated by zinc transporters that act to absorb diet-derived zinc for distribution to the peripheral tissues[321]. But excess zinc is toxic to both intestinal epithelial cells[308,315]and also peripheral tissue cells, thus zinc transporters maintain cytosolic zinc levels via both influx and efflux of zinc ions[306]. The physiological role of zinc and zinc transporters have been best studied in the intestinal tract, where zinc is required for intestinal epithelial homeostasis, a process mediated by several zinc transporters. In intestinal absorptive enterocytes, ZIP4 and ZnT1 regulate zinc absorption, while ZIP5 and ZnT5B regulate zinc excretion. In intestinal Paneth cells, ZnT2 and ZIP4 are required for zinc accumulation in Paneth cell granules and are important for Paneth cell maintenance overall. Of potential importance to intestinal cancer, ZIP4 and ZIP7 are expressed in the intestinal crypt stem cell compartment where they contribute to Lgr5+ stem cell maintenance and help maintain transit amplifying cell proliferation[312].
Zinc transporters and GI cancer
The role of zinc transporters in human cancer have been best studied in prostate and breast cancer[308,322]. In the prostate gland ZIPs, especially ZIP1, are considered tumor suppressors[323]. In breast cancer, increased levels of zinc correspond to upregulation of several ZIPs (and a few ZnTs as well), thus zinc transporters as a group are considered as oncogenes in breast cancer[324]. In the GI tract, the majority of reports show upregulation of both ZIPs and ZnTs in all major GI organ cancers (Table 5),although for some cancers there is clearly conflicting evidence. The strongest evidence for dysregulation of zinc transporters in GI tract cancers has been for PC, where zinc transporter upregulation has been associated with enhanced cancer cell migration and worse patient prognosis. This is especially true for ZIP4. In one study increased mRNA expression of ZIP4 was observed in 16 of 17 pancreatic adenocarcinoma samples[325], a finding that was supported in ZIP4-expressing xenografts in mice that yielded larger tumors[326]. These findings were confirmed by several other groups[327-329], such as Xu et al[329]who found that ZIP4 was upregulated in a set of 23 PCs and that ZIP4 expression could be used as a diagnostic and prognostic marker.While the case of ZIP4 as an oncogene is very clear, for other zinc transporters the data is sometimes contradictory, with some reports of upregulation and others of down-regulation of non-ZIP4 zinc transporters[313,330]. Studies employing zinc-sensitive histochemical staining have reported a general reduction in zinc levels in PC samples[331,332], a report that is difficult to reconcile with upregulation of zinc transporters, although it has also been noted that increases in zinc transporter mRNAs do not always correspond to increased protein expression and activity as many zinc transporters, including ZIP4, are regulated by posttranslational mechanisms[322].Further, some studies have shown down-regulation of virtually all zinc transporters in PC, with the exception of ZIP4[313]. Another case for zinc transporters acting as an oncogene or tumor suppressor is in EC, where an inherited genetic variant in the 5'untranslated region of ZIP6 results in constitutive expression of ZIP6, enhancing the malignancy of ESCC cells[333]. In ESCC cell lines upregulation of ZIP6 enhances proliferation, migration, and invasion of cancer cells, while down-regulation of ZIP6 suppresses these effects[333]. Similarly, Jin et al[334]reported that knockdown of ZIP5 significantly inhibited human ESCC cell progression and Kumar et al[335]reported upregulation of ZIP7 in ESCC. In contrast, other studies have reported that zinc deficiency contributes to progression of EC. Choi et al[336]reported that zinc supplementation inhibited proliferation of ESCC cell lines, a process mediated by inhibition of Orai-mediated SOCE and subsequent Ca2+oscillations. For other GI cancers such as CRC and GC, there is less evidence but those reports point to an oncogenic role for zinc transporters[337-339], despite zinc's known protective effects against colonic inflammation.
Mechanisms in cancer cells
The mechanisms underlying the effects of dysregulation of zinc homeostasis and zinc transporter expression in cancer are unclear, and are almost certainly tissue and cancer specific as seen in the evidence that zinc transporters appear to clearly act as tumor suppressors in the prostate gland but as oncogenes in breast cancer. Less is known in GI tract cancers, and that data is controversial (see conflicting PC and EC studies above) but some potential mechanisms of action are listed below.
Zinc signaling within cells:Changes in intracellular concentration can lead to it acting as a second messenger for external signals, including activation of mitogenactivated protein kinase (MAPK), extra-cellular signal-related kinase (ERK) and the c-Jun N-terminal kinase (JNK) pathways that can act to phosphorylate proteins involved in regulating cell proliferation, differentiation and apoptosis[306,322]. This has been best studied in breast cancer where ZIP-mediated signaling pathways promote cell proliferation and metastasis in luminal and tamoxifen-resistant breast cancer cells[322]. Overall, zinc is recognized as an important signaling molecule in normal cell functions as well as pathologies such as cancer. How zinc signaling is conveyed from zinc transporters to downstream signaling pathways is still largely unclear.
Regulation of the intestinal stem cell compartment:In normal tissue several zinc transporters are involved in maintenance of Lgr5+ stem cells and Paneth cells, which themselves are important for stem cell maintenance[312]. Further evidence for this mechanism is the role of ZIP7 in resolving ER stress in the intestinal stem cell compartment[340]. Mice lacking ZIP7 in intestinal epithelium triggered ER stress that led to loss of intestinal stem cells and proliferative progenitor cells.
Requirement for zinc in rapidly proliferating cells:Zinc is required for protein structure, catalytic activity, such as for zinc finger transcription factors.
Action of increased cellular zinc on other ion channels:TRP Ca2+and K+channels are sensitive to zinc activation[308].Zinc deficiency: Zinc deficiency leading to disruption of intestinal epithelial barrier function and enhanced colonic inflammation as zinc supplementation has been shown to improve inflammatory bowel disease-related phenotypes in animal models.
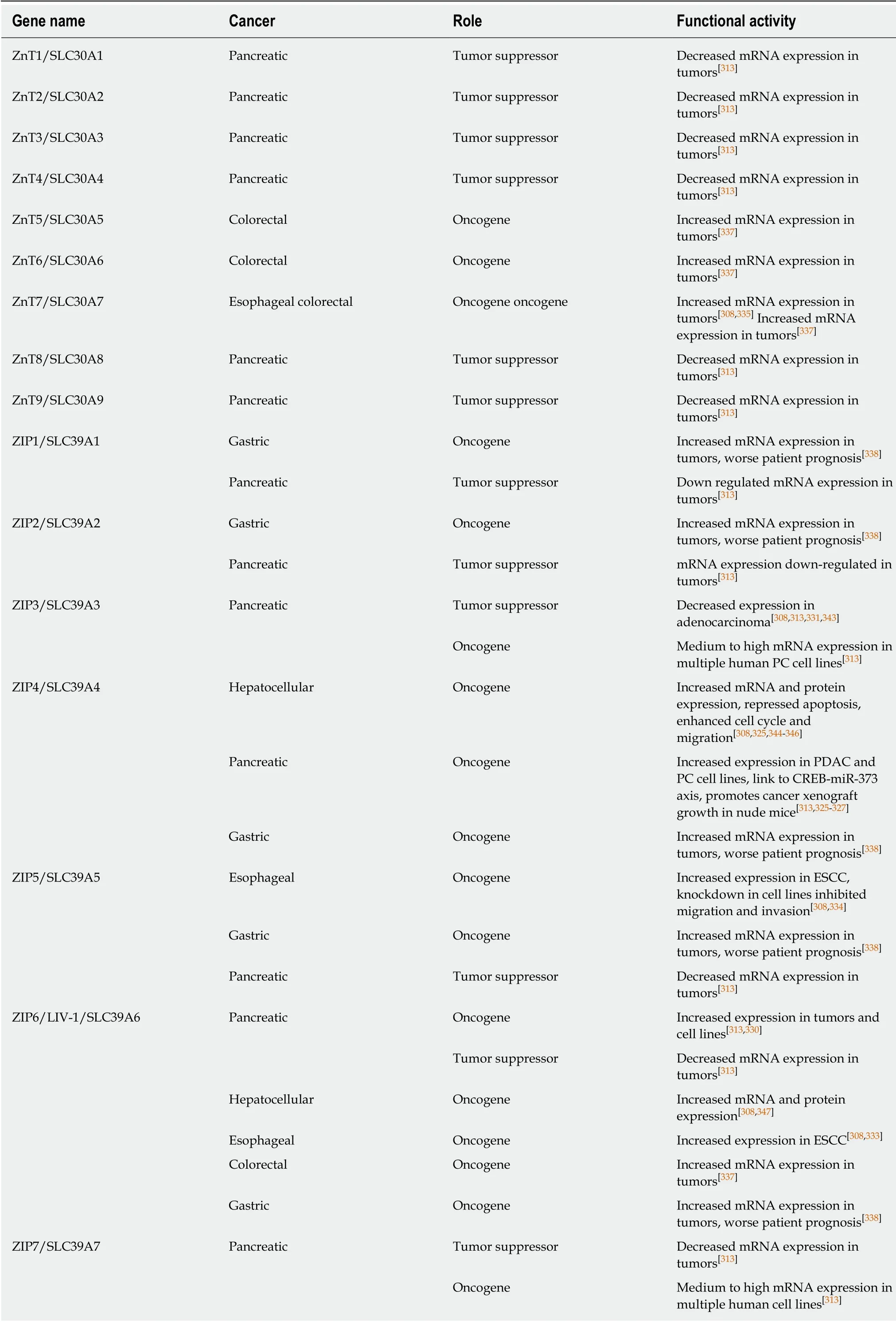
Table 5 Zinc transporters
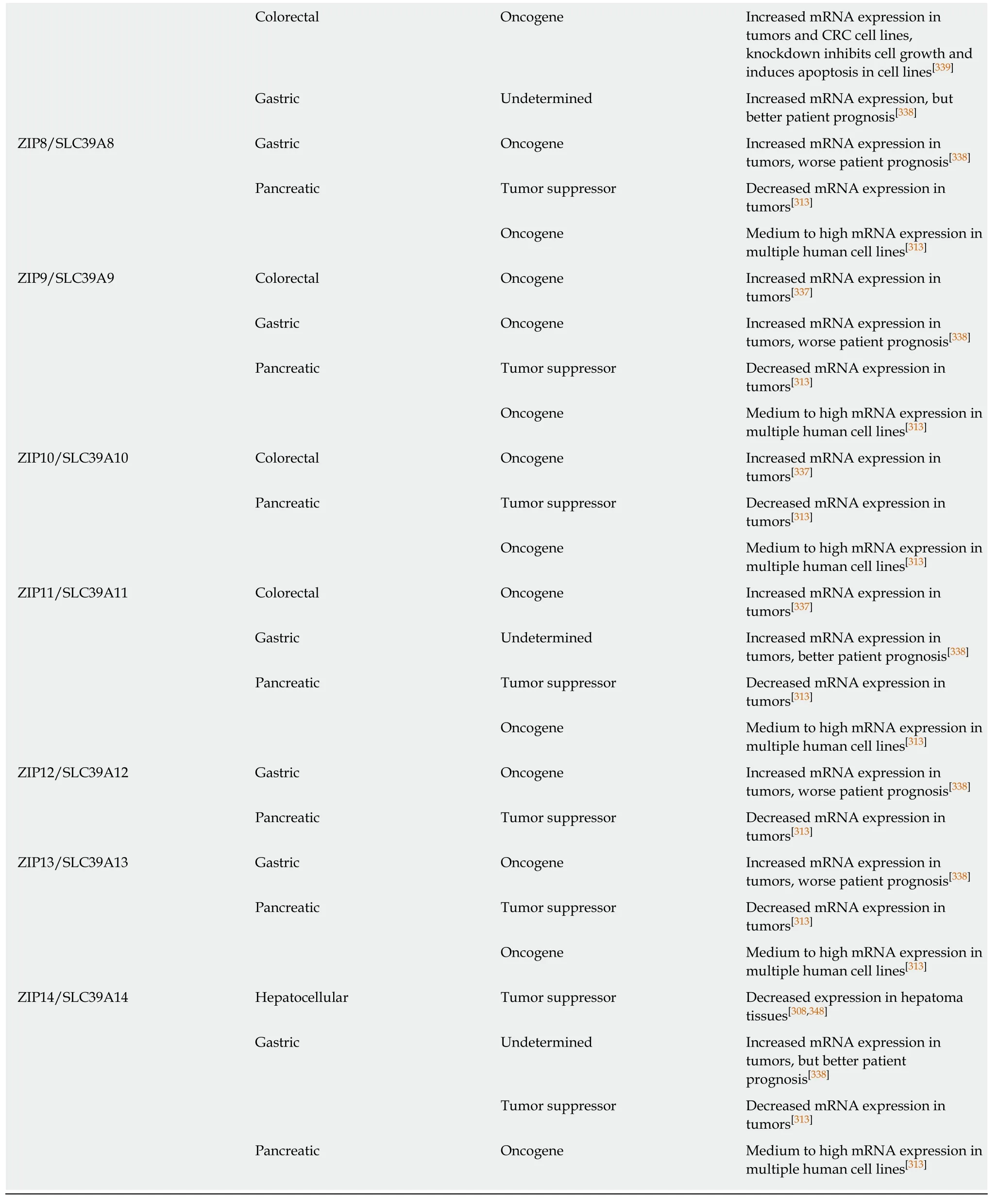
PDAC: Pancreatic ductal adenocarcinoma; ESCC: Esophageal squamous cell carcinoma; CRC: Colorectal cancer; PC: Pancreatic cancer.
Therapeutic opportunities
In contrast with other ion channels discussed in this review drugs targeting various zinc transporters have been slow in development. Myers[341]and Bin et al[342]discuss some of the challenges to drug development represented by zinc transporter structure. One of these, ZnT8, which is especially associated with type-2 diabetes is the subject of current drug development research. Potentially, knowledge gained from targeting ZnT8 could lead to targeting of other transporters such as ZIP4 in PC. One therapeutic area that is immediately available is zinc supplementation, also discussed by Myers and Bin et al[342]. For conditions, including cancers, that demonstrate zinc deficiency, zinc supplementation may have immediate therapeutic value. This has been demonstrated for inflammatory bowel disease and may have utility for zincdeficient cancers. See Table 5 for a full listing of zinc transporters and their role in GI cancers.
CONCLUSION
Ion channels play an essential role in the GI tract, mediating a range of cellular and tissue processes. They are also commonly dysregulated in major GI malignancies such as CRC, HCC, PC, GC and EC. In these cancers, ion channels modulate many of the well-known hallmarks of cancer, with increasing evidence that ion channels are important for the regulation of tissue and cancer stem cells. The influence of ion channels on cancer cell processes has led to cancer being described as a channelopathy. For a summary of mechanisms of selected ion channels in GI cancer see Figure 3. Notably, ion channels represent potentially important clinical targets for several reasons.
First, ion channels and transporters are predominantly found in the PM of the lumens of GI tract organs thus the majority of ion channels should be accessible to therapeutic drugs.
Second, the structures of all of the major families of ion channels found in the GI tract are known and their functions have been well-characterized, primarily due to studies prompted by dysregulation of these ion channels outside the GI tract, e.g.,CFTR in CF lung pathology, thus they should be readily amenable to new drug design and preclinical and clinical testing.
Third, drugs are currently used to target several ion channels for disorders outside the GI tract e.g., retigabine for KCNQ-deficiency and many other examples, thus these drugs can be repurposed for clinical use in the GI tract. Current examples of drug repurposing in the GI tract include CFTR potentiators, activators, correctors, and amplifiers such as for patients with specific CFTR mutations. These include Ivacaftor(potentiator for > 25 CF-causing mutations, including G551D), and Tezacaftor and Lumacaftor (correctors for patients with F508delta mutations which account for aaproximately 70% of CF patients). These drugs have been shown to be effective in ameliorating lung pathology in CF patients and are now being used to treat several GI pathologies in CF patients. For example, Ivacaftor is being used to treat pancreatitis,and intestinal inflammation (including producing an improvement in gut microbiota)and Lumacaftor has been shown to improve intestinal bicarbonate permeability. As CF patients are highly susceptible to the development of CRC, these drugs may be readily repurposed for prevention and treatment of CRC, likely in combination with other standard therapy. Data generated by the repurposing efforts underway for noncancer therapy will provide support for the next step into the cancer clinic.
Fourth, further research into the mechanisms of action of various ion channels,including the rapidly growing utility of bioinformatics analysis, can lead to greater drug repurposing strategies. For example, through a bioinformatics approach,tricyclic antidepressants were recently and rapidly repurposed by the FDA for use in treatment of small cell lung carcinoma[349]. In other cases, research into mechanisms of drug action reveals ion channels as novel mediators. For example, it has long been known that daily aspirin is protective against CRC, but only recently has it been determined that the mechanism of this effect is likely through the remodeling of the SOCE Ca2+channel[256]. Further understanding of the canonical and non-canonical roles that ion channels play in cancer development can lead to further repurposing of drugs to treat specific GI cancers[350]. This is especially true as precision cancer medicine embraces combinatorial treatment modalities.
Fifth, ion channels and transporters can provide novel cancer biomarkers with diagnostic and prognostic implications, e.g., KCNQ1 in later stage CRC, where patients who maintain high expression of KCNQ1 show better disease free survival at stage II and III CRC[38], and a 23-month survival advantage for stage IV CRC patients following hepatic resection[37]; CFTR in CF-related CRC[122-126]; hERG1 in several GI cancers[59-74]. and zinc transporters such as ZIP4 in PC[325-329]. For example in CRC,hERG1 is not expressed in normal colon mucosa but is upregulated in adenocarcinoma, with its highest expression in CRC metastasis. It is noted that stereoselective hERG1 channel blockers for treatment of cardiac arrhythmias have been used in the clinic for several decades.

Figure 3 Oncogenic mechanisms of selected ion channels. Because ion channels influence the basic biochemical environment of the cell as well as complex interactions with other proteins, they have profound and pleiotropic effects on cell function. As a result, it is often difficult to determine specific mechanisms for oncogenic phenotypes. However, progress has been made in defining mechanisms in some cases. This figure shows examples from each category of channels with accompanying pathways linking dysregulation of channel function to tumorigenesis. For additional information and references please see text and Tables 1-5. GI:Gastrointestinal; EMT: Epithelial to mesenchymal transition; TRP: Transient receptor potential; SOCE: Store-operated calcium entry; VGSC: Voltage-gated sodium channels; STIM1: Stromal interaction protein 1.
Sixth, there is research data linking several ion channels to regulation of the stem cell compartment in GI tract organs. This is especially true for the intestinal tract. For example, both CFTR[93,112]and KCNQ1 (R.Cormier and P.Scott, unpublished studies)are expressed in the stem cell compartment of the colon, where they have been shown to influence stem call capacity in mouse organoids models[37,128], as well as regulating the expression of stem cell related genes, again in transgenic mouse models[37,128]. As the intestinal stem cell is thought to be the precursor for the intestinal cancer stem cell,better understanding of the underlying mechanisms of how ion channels such as CFTR and KCNQ1 may regulate stem cell function will be very important. A key tool in this research and a technical advance in therapeutic development has been the creation of patient-derived tissue and cancer organoid surrogate models[136], e.g., that have been used to test the function of CFTR[137,138], as well as the efficacy of CFTR modulator drugs in CF patient-derived rectal organoids[208]. The same strategy of biobanking of patient-derived organoids is currently used to test patient-specific CRC treatment protocols that can include ion channel modulator drugs.
Seventh, as patient genomic sequencing efforts increase, more has become known about specific germline genomic variants in ion channel genes and potential susceptibility to GI tract cancers. For example, CF patients who are homozygous for CFTR mutations are at a significantly heightened risk for developing early onset CRC.What is the risk for CRC for heterozygous carriers of CFTR mutations, a group that represents more than 10 million individuals in the United States alone? CRC patients whose cancers show reduced expression of CFTR demonstrate worse CRC disease prognosis[128]. Current ongoing studies of CRC risk in CFTR carriers may help inform on the lifetime risk of CRC in these patients, potentially leading to earlier screening and chemoprevention.
A final comment here, as discussed by Humphries et al[351], is that while there exists great promise in targeting ion channels in human diseases there remains significant challenges, especially related to target specificity and off-target toxicity, before development of ion channel targeting can lead to more widespread effective personalized cancer treatment.
杂志排行

World Journal of Gastroenterology的其它文章
- Targeted therapies in metastatic gastric cancer: Current knowledge and future perspective
- lncRNA-SNHG15 accelerates the development of hepatocellular carcinoma by targeting miR-490-3p/histone deacetylase 2 axis
- Sirtuin 1 alleviates endoplasmic reticulum stress-mediated apoptosis of intestinal epithelial cells in ulcerative colitis
- Up-regulated Wnt1-inducible signaling pathway protein 1 correlates with poor prognosis and drug resistance by reducing DNA repair in gastric cancer
- Hepatitis C virus clearance and less liver damage in patients with high cholesterol, low-density lipoprotein cholesterol and APOE ε4 allele
- Nomogram to predict prolonged postoperative ileus after gastrectomy in gastric cancer