Al原子在Si表面扩散动力学的第一性原理研究*
2019-10-25张恒黄燕石旺舟周孝好陈效双
张恒 黄燕 石旺舟 周孝好† 陈效双
1) (中国科学院上海技术物理研究所,红外物理国家重点实验室,上海 200083)
2) (上海师范大学数理学院,上海 200234)
为了更好地在Si衬底上外延生长GaN薄膜,需要先生长缓冲层(如AlN),其中能否对起始的金属Al层实现可控生长,将决定最终外延层的材料质量.本文采用基于密度泛函理论的第一性原理计算,理论上模拟计算了金属Al原子分别在清洁的、H原子和Cl原子钝化的Si(100)及Si(111)表面的吸附及扩散动力学行为.研究结果显示,在清洁的Si(100)表面上,Al原子易于吸附在沟槽中Tr位点,沿沟槽呈曲折状扩散; 在H钝化、Cl钝化的Si(100)表面上,Al原子易于吸附在二聚体列顶部的H位置,在二聚体列顶部沿直线扩散.在不同方式处理的Si(111)表面,Al原子的最稳定吸附位置相同,均易吸附于第二层Si原子的Top位(T4位点),扩散路径类似,均沿T4到H3 (空心位点)的路径扩散.无论是Si(100)还是Si(111)表面,H钝化、Cl钝化处理Si表面均有效降低Al原子的扩散能垒,使Al原子更容易在二维表面上扩散,并通过吸附能的比较以及差分电荷密度图分析,讨论了不同Si表面状态对金属Al原子吸附和扩散行为调制的物理机制.
1 引 言
GaN是发光二极管、高频电子仪器、紫外波段探测器等半导体设备常用的材料,具有宽带隙(Eg=3.4 eV)、耐腐蚀等优点[1,2].GaN需要外延生长在异质结衬底上,其中蓝宝石(Al2O3)、碳化硅(SiC)和Si是常用的衬底[3,4].蓝宝石由于稳定性高而不能进行微加工,碳化硅因为成本太高无法大面积应用[5].与这些衬底相比,Si具有价格低、导热性好和技术成熟的优点,因此被认为是一种最有前景的GaN外延衬底[6].然而,Si和GaN之间存在晶格失配(20%)和热膨胀系数失配(56%)的问题,阻碍了Si衬底上GaN外延生长[7].因此需要在Si衬底上先生长适当的缓冲层(如AlN)来实现Si基GaN的高质量生长[6].最近实验报道在生长AlN缓冲层之前,通过控制预铺纯金属Al层的生长时间和温度,可以使GaN晶体质量和表面形态得到改善[8-11].由于外延生长过程中很难在实验上对其微观反应机理进行表征,因此,为了更好地实现对Si基GaN材料的可控外延生长,需要结合理论模拟等手段,从原子尺度来理解该生长过程的微观机制.近年来,在二维材料研究中,例如,如何进一步拓展其功能的应用,也需要从原子尺度去理解和认识其中的微观机理[12-14].
借助于理论模拟计算,可以分析预铺Al原子在Si表面的吸附几何位置、成键和电子转移等情况.例如Albao等[15]计算Al原子易于吸附在Si(100)沟槽中的Tr位点,在Si(100)表面Al原子存在5条扩散路径,在垂直和平行于Si-Si二聚体对应的最低扩散能垒分别是E⊥=0.466 eV,E∥=0.305 eV.Luniakov等[16]报道在Si(111)表面Al原子的稳定吸附位置为T4位点,微动弹性带(nudged elastic band,NEB)法优化的扩散路径是绕顶部Si原子,从T4位点到相邻T4′,扩散能垒约为3.1 eV.另外,Matsuo等[17]研究发现,对Si(111)进行H钝化处理后,Ga和Al原子的吸附能会随着H覆盖率的降低而增加,表明H钝化能够抑制Si表面原子的吸附能.上述研究清楚地表明不同的Si表面状态,将会显著地影响其表面吸附原子的稳定构型以及扩散动力学等行为.在本研究中,基于密度泛函理论(density functional theory,DFT)的第一性原理计算,我们详细地研究了Al原子在不同的Si表面,包括清洁、H原子钝化、Cl原子钝化的Si(100)和Si(111)表面的吸附和扩散动力学行为,并通过吸附能、差分电荷密度分析了其背后的物理缘由.该研究结果可以为实验上优化Si基外延生长GaN材料提供理论参考.
2 计算方法
本文计算采用基于密度泛函理论的第一性原理商业软件VASP[18,19].首先沿着(100)和(111)方向分别建立Si(100)和Si(111)表面.这两种表面均含有七层Si原子,底部Si原子的悬挂键被H原子钝化.H原子和底部四层Si原子被固定,其余三层Si原子可以自由移动.Si表面设置大于10 Å的真空层,以防止相邻表面之间相互作用.交换关联能采用Perdew-Burke-Ernzerhorf形式的广义梯度近似(GGA)[20].选用的布里渊区采用 (5×5×1)的K点取样,平面波截断能为350 eV.静态计算时的收敛标准为自洽迭代的能量差小于1.0×10-4eV.在原子结构弛豫过程中,收敛标准为原子所受的力小于0.01 eV/Å.收敛条件满足后,结构优化完成.采用NEB方法搜索最小能量路径和确定过渡态结构[21-23].该方法在确定的始末两态之间路引入一些过渡构型,通过结构弛豫,得到扩散的最小能量路径并计算出在已知扩散途径之间的扩散能垒.
通过计算Al原子的吸附能来表示其在Si表面的稳定性,定义公式如下:
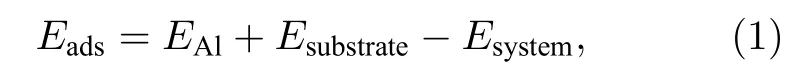
其中,EAl表示Al原子吸附前的能量,Esubstrate表示吸附前Si衬底的能量,Esystem表示Si衬底吸附Al原子稳定后系统的总能量,Eads为Al原子在Si表面的吸附能.吸附能如果是正值,表示吸附过程稳定.吸附能越大,吸附越稳定.反之吸附能如果是负值,表示该吸附过程不稳定,不能自发进行.
3 结果与讨论
3.1 Al原子在清洁Si(100)表面的吸附
在Si(100)表面,Si原子与相邻的Si原子共用一对电子,形成稳定的非对称二聚体,此时体系能量最低.如图1所示在清洁Si(100)表面会呈现(2×2)的重构表面,Si-Si二聚体处于同一高度呈交叉排布,测量二聚体中Si-Si键的键长是2.38 Å,二聚体的倾斜角度是18°,计算结果与文献[24]报道一致.对Si(100)表面的悬挂键用H原子或者Cl原子钝化后,Si-Si二聚体由交叉排列变成平行排列[25,26].为了方便观察Si表面二聚体排列变化,我们分别用深蓝色和红色标记二聚体中较高和较低两种位置的Si原子.通过对Si(100)表面Si原子的对称性分析,我们选择Tr点(沟槽部分第四层Si原子顶部)、T点(第三层Si原子顶部)、M点(二聚体列中Si原子的中间部分)、H点(二聚体列的中心空位点)以及B点(Si-Si二聚体的桥位)作为Al原子的吸附位置,对比研究了清洁的Si(100)表面和H钝化、Cl钝化Si(100)表面后,Al原子在这几个位置的吸附以及扩散情况.
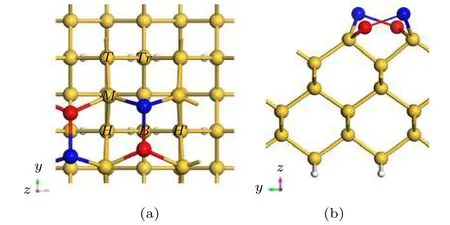
图1 Si(100)表面的几何结构,其中深蓝色和红色用于标记Si-Si二聚体中高和低两种位置的Si原子 (a)俯视图;(b)侧视图Fig.1.The geometry of the Si(100) surface,in which dark blue and red are used to mark the Si atoms in the high and low positions in the Si-Si dimer:(a) Top view; (b) side view.
经计算Al原子在清洁的Si(100)表面能够稳定吸附在Tr点、M点和H点(表1),对应的吸附能分别为4.01,3.96,3.63 eV.其中Tr位点为Al原子的最稳定吸附位点.如图2(a)和图2(b)左图所示,吸附在Tr位点的Al原子与相邻的两个Si-Si二聚体的Si原子成键,Al-Si键长分别均为2.45 Å.形成的Al-Si键导致Si-Si二聚体中Si原子间距离延长并发生断裂 (距离为2.77 Å),同时位于低位的Si原子(红色)升高,与高位的Si原子(深蓝色)水平.从差分电荷密度图 (图2(c)左图)同样可以看出,Al原子周围为红色区域表现为Al原子失去电子,与Al原子成键的Si原子得到电子,表明二者能够形成极性共价键.
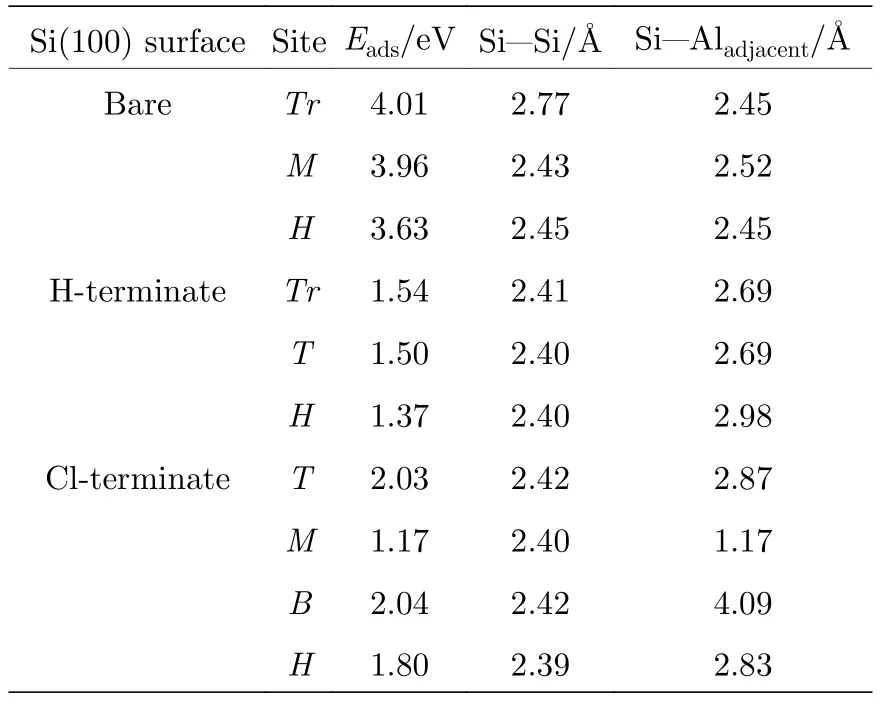
表1 Al原子吸附在清洁、H钝化和Cl钝化Si(100)表面的吸附能Ead(eV)和结构参数; 其中Si-Si为二聚体Si-Si的键长,Si-Aladjacent为Al原子与邻近Si原子之间的距离Table 1.Adsorption energy Ead (eV) and structural parameters of Al atom adsorbed on clean,H-terminate and Cl-terminate Si(100) surface; Si-Si is the bond length of dimer Si-Si,Si-Aladjacent is the distance between the Al atom and the adjacent Si atom.
如图2(a)和图2(b)中图所示,当Si(100)表面的Si原子被H原子或者Cl原子钝化以后,发现表面的Si-Si二聚体会变成同一高度,并且平行排列.测量二聚体中Si-Si键长分别为2.42 Å和2.44 Å,比重构的清洁Si表面的二聚体键长分别缩了0.04 Å和0.06 Å.说明H钝化或Cl钝化Si(100)表面能够有效延长二聚体中Si-Si键长.
我们将Al原子放在H钝化Si(100)表面,计算Al原子能够稳定吸附在Tr,T和H位点(表1),对应的吸附能分别为1.54,1.50,1.37 eV,吸附能与Al原子在清洁的Si(100)表面稳定吸附位点的吸附能相比显著降低.为了研究H钝化Si(100)表面对Al原子与Si原子相互作用的影响,我们选择靠近钝化层的H位点进行研究.当Al原子稳定吸附在H位点时,测量二聚体中Si-Si键长为2.40 Å,比吸附Al原子前键长短0.02 Å.从差分电荷密度图 (图2(c)中图)同样可以看出,Al原子失去电子,但周围红色区域明显少于在清洁的Si(100)表面,说明Al失电子减少,Al原子和Si原子相互间作用减弱.
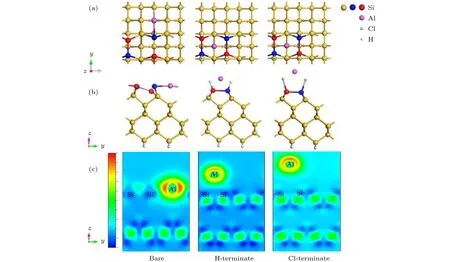
图2 Al原子吸附在清洁Si(100) Tr位点、H钝化Si(100) H位点和Cl钝化Si(100) 表面H位点 (a)俯视图; (b)侧视图; (c)差分电荷密度图Fig.2.Al atom adsorption on the clean Si (100) Tr site,H-terminate Si (100) H site and Cl-terminate Si (100) surface H site:(a) Top view; (b) side view; (c) differential charge image.
同样地,我们将Al原子放在Cl钝化Si(100)表面,计算Al原子能够稳定吸附位点为T,M,B,H四个位点(表1),对应的吸附能分别是2.03,1.17,2.04,1.80 eV.同样选择靠近钝化层的H位点进行研究.当Al原子稳定吸附在H位点时,二聚体中Si-Si键长为2.39 Å,与Al原子吸附前比,键长缩短0.05 Å.同时形成的Si-Cl键长为2.07 Å,比Si-H键长0.57 Å,长的Si-Cl键使Al原子远离表面的Si原子.从差分电荷密度图 (图2(a)和图2(b)右图)同样可以看出,Al原子位置明显高于在清洁的和H钝化Si(100)表面的位置,但失电子转移情况却在二者之间,是由于Cl原子电负性强能够吸引Al原子失去更多的电子所致.这些结果说明Cl钝化Si(100)表面能够有效弱化Al原子与表面Si原子间的相互作用.
为了更好地理解金属Al原子在清洁、氢化、氯化Si(100)相互作用,我们分别计算了三种Al原子在Si(100)表面稳定吸附Al原子的Bader电荷转移,如图3所示,其中Al原子在清洁、H化、Cl化Si(100)表面吸附的位点分别为Tr,H,H位置.
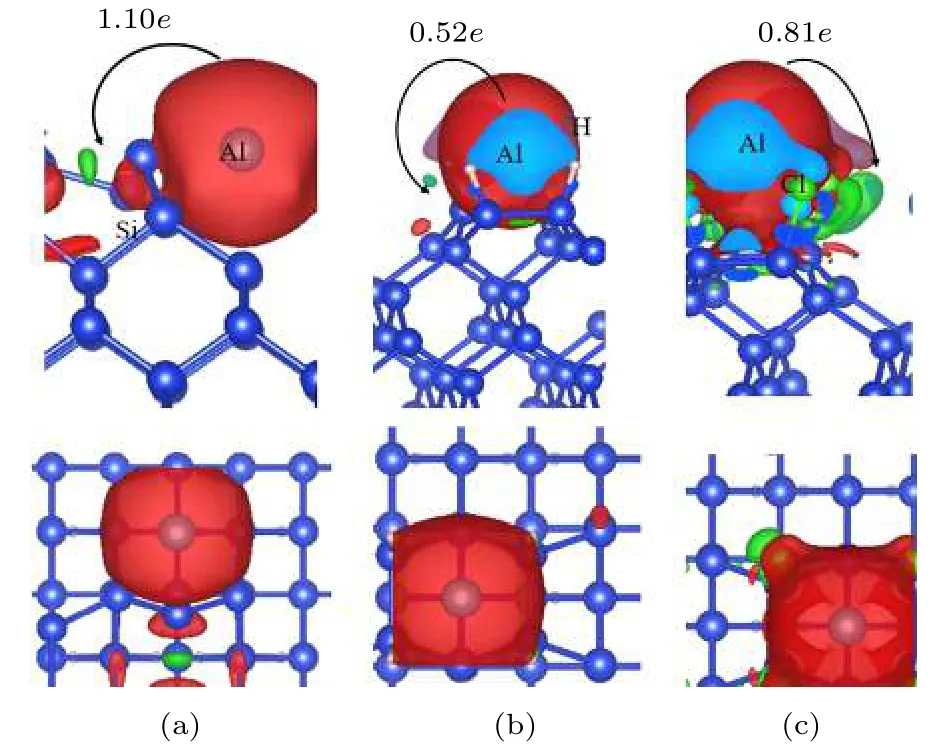
图3 Al原子吸附在(a)清洁、(b)氢化、(c)氯化Si(100)表面后差分电荷图和Bader电子转移情况Fig.3.Bader charge transfer for Al atom adsorption on (a)the clean Si (100) Tr site,(b) H-terminate Si (100) H site and (c) Cl-terminate Si (100) surface H site.
图3中红色区域表示电子耗散区,绿色区域代表电子聚集区,在清洁Si(100)表面的Al原子会转移1.10e给邻近的Si原子,表示Al原子与Si(100)表面的Si原子发生了强烈的相互作用,同时也印证了正是由于Al与二聚体当中Si的相互作用使得二聚体中的Si-Si键断裂.当表面被H钝化或者Cl钝化以后,由于Al与Si原子的距离相对变长,使得Al与Si之间的相互作用更弱,相当于H或者Al原子对Si衬底起了一定的保护作用,对应H化和Cl化Si(100)表面,Al原子分别转移了0.52e和0.81e.与Al原子在H化或Cl化Si表面的吸附能的大小变化非常相符,对应的吸附能分别是1.37 eV和1.80 eV.正是因为Al原子在Cl化Si(100)表面转移的电子多,释放的能量也就越多,但仍然比清洁Si(100)表面的吸附能要小,转移的电子要少,即H或者Cl化Si(100)表面有效地减弱了Al与Si之间的相互作用.Al原子吸附在清洁、H化、Cl化Si(100)表面时,都会把电子转移给邻近的Si原子.从几何位置上来看Al原子在H化、Cl化Si(100)表面吸附的稳态位置相同,而Al原子在清洁Si(100)表面吸附时,会稳定吸附在沟槽的位置.
3.2 Al原子在Si(100)表面的扩散
如图4(a)所示,在清洁的Si(100)表面Al原子是从沟槽中的Tr位点绕相邻二聚体中的Si原子弯曲扩散到M位点,即Al原子绕着相连Si原子顺时针移动了1/4的圆.Al原子在扩散路径上的竖直高度一直处于上升的状态,吸附在M位点的Al原子高度比吸附在初始位置Tr位点升高0.039 Å.而在H钝化和Cl钝化Si(100)表面Al原子扩散的路径为从二聚体的空心位置H位点沿直线扩散到相邻对称位置的H'位点.Al原子在扩散路径上的竖直高度出现先升高后降低的趋势,吸附在鞍点时竖直高度最高,分别比吸附在初始位置H位点高0.025 Å和0.018 Å.图4(b)是Al原子在Si(100)表面扩散的能垒图,黑线是Al原子在清洁的Si(100)表面的扩散.计算发现Al原子无法稳定吸附在沟槽中的T位点,由于Al原子从沟槽中的Tr位点扩散到对称的Tr' 位点需要克服巨大的势垒,因此Al原子最不容易吸附在沟槽中的T位点,即Al原子扩散时鞍点位于远离T位点附近的空心位.红线和蓝线分别是Al原子在H钝化和Cl钝化Si(100)表面扩散的能垒图,鞍点位于H钝化或Cl钝化Si(100)表面的B位点.有趣的是,我们发现Al原子会吸附在Cl钝化Si(100)表面B位点,而B位点却是Al原子在清洁的和H钝化Si(100)表面的不稳定位点.分析原因是Si(100)被Cl处理后,形成Si-C比Si-H键长,形成的Si-Cl键为极性共价键,对Al原子有强的吸引作用所致.从表3可知,Al原子在清洁、H钝化、Cl钝化Si(100)表面扩散的能垒分别为0.60,0.38,0.13 eV.很明显在Si(100)表面的Si原子被H或者Cl钝化以后,表面的扩散能垒显著降低.特别是Al原子在Cl原子钝化Si(100)表面上扩散,扩散能垒比清洁的Si(100)表面降低了0.47 eV.同时扩散路径也表明,在Si(100)表面,Si原子被H或者Cl钝化以后,改变了Al原子在Si(100)表面的扩散路径.扩散路径都是从表面的H点(空位点)扩散到相邻的空位点,而不再是在沟槽部分进行扩散.这使得Al原子的扩散变得更加容易,有利于在Si基GaN的实际生产中预铺平整的Al原子.
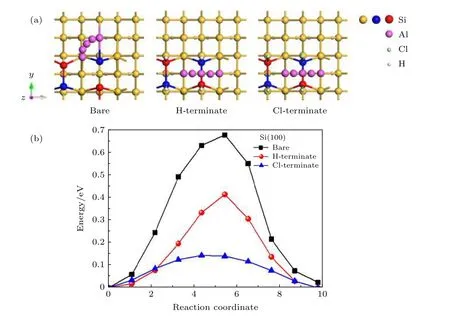
图4 Al原子在清洁、H钝化、Cl钝化Si(100)表面 (a)扩散路径和; (b)扩散能垒Fig.4.Al atoms on clean,H-terminate and Cl-terminate Si(100) surfaces:(a) Diffusion paths; (b) diffusion barriers.
3.3 Al原子在Si(111)表面的吸附
在Si(100)表面,由于Si-Si二聚体的形成会导致Si表面重构,但是在Si(111)不会出现这种现象.在Si(111) (2×2)表面每一个Si原子都有一个悬挂键,会与金属Al原子产生强烈的相互作用.为了饱和悬挂键,我们分别用H或者Cl钝化Si(111)表面.如图5所示,为了方便观察Al原子与表面Si原子成键情况,我们分别用浅蓝色和粉红色标记第二层和第四层Si原子.通过几何对称性分析,我们选择T1(第一层Si原子的top位置)、B2(Si-Si键的桥位)、H3(表面Si的六元环的空位部分) 以及T4(第二层Si原子的top位置)几个高对称点作为Al原子的吸附位置,对比研究了清洁的Si(111)表面和H钝化、Cl钝化Si(111)表面后,Al原子在这几个位置的吸附以及扩散情况.
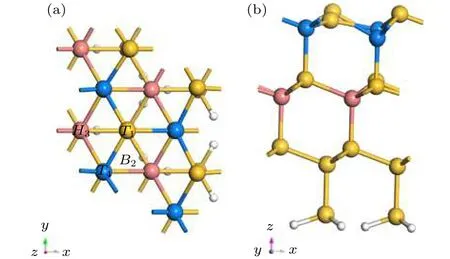
图5 (a)和(b)分别为清洁Si(111)表面俯视图和侧视图T1,B2,H3和T4为Al原子在Si(111)表面的吸附的四个高对称位Fig.5.(a) and (b) are the top and side views of the clean Si(111) surface,respectively; T1,B2,H3 and T4 are the four highly symmetric sites of adsorption of Al atoms on the Si(111) surface.
计算结果显示如表2所列,Al原子在清洁的Si(111)表面,最稳定的吸附位置在第二层Si原子的top位置(T4位点),与Luniakov等[15]报道的最稳定位置一致,对应的吸附能最大是4.51 eV,H3位点是亚稳态吸附位点,对应的吸附能是4.35 eV.在T4位点,Al原子会饱和掉三个位于T1位点Si原子的悬挂键,形成键长为2.55 Å的化学键,数据与文献[13]报道一致.从图6(c)左图的差分电荷密度可以看出,Al原子失去电子,邻近Si原子得到电子.
当表面第一层Si原子被H和Cl处理后,Si(111)表面的Si原子的悬挂键被H原子或Cl原子饱和掉.从表2可知,Al原子在H钝化和Cl钝化Si(111)表面的最稳定吸附位置与清洁的Si(111)表面相同,都在T4位点,对应的吸附能分别是1.36 eV和2.03 eV.亚稳态吸附位点均为H3位点,对应的吸附能分别为1.13 eV和1.99 eV.由于H或者Cl处理Si表面后钝化了Si(111)表面的悬挂键后,Al原子与第一层Si原子的距离相对清洁的Si(111)表面延长了0.54 Å和1.72 Å,竖直方向的高度相对升高了0.14 Å和1.81 Å,因此Al原子不能与表面邻近的Si原子成键.从图6(c)中图的差分电荷密度可以看出,H钝化的Si(111)表面,Al原子失电子转移给了邻近的Si原子,但是相对于清洁的Si(111)表面,电子转移的数量减少了,弱化了Al与Si之间的相互作用; 在Cl钝化的Si(111)表面,由于Cl原子的电负性强,Al原子会转移一部分电子给Cl原子,因此转移的电子会比吸附在H钝化Si(111)表面的电子多.这些结果表明,在Si(111)表面做H或Cl的钝化处理可以弱化Si和Al原子之间的相互作用,有利于Al原子在Si表面的二维扩散.这也证实了Matsuo等[16]研究发现H钝化能够抑制Si(111)表面原子吸附能的结果.
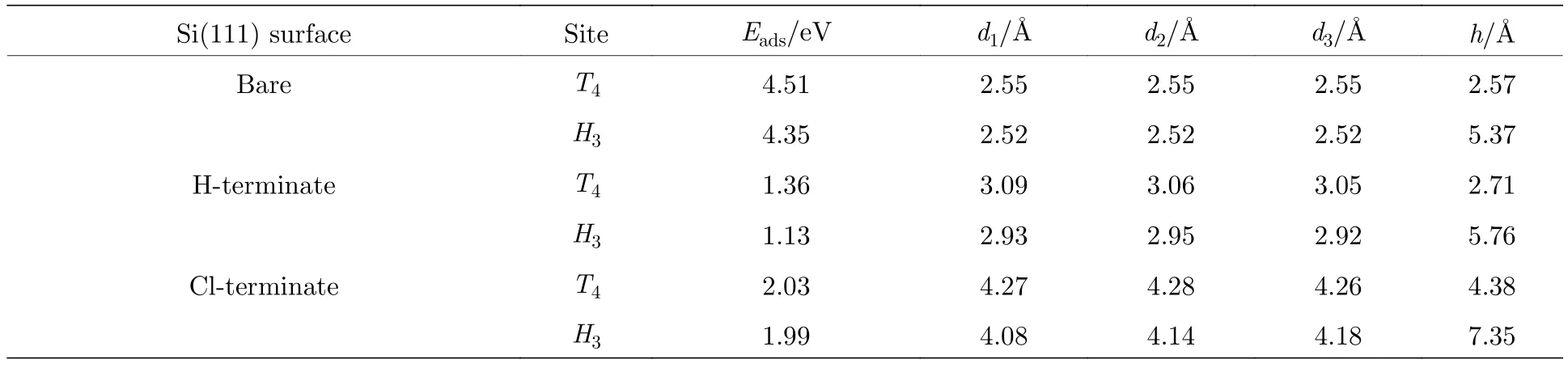
表2 Al原子吸附在清洁、H钝化、Cl钝化Si(111)的几何位点,其中Eads为对应位点的吸附能,d1,d2,d3为Al原子与邻近三个Si原子之间的距离,h为竖直方向Al原子与邻近Si原子之间的距离Table 2.Al atom adsorption in the clean,H-terminate,Cl-terminate Si (111) geometric site,where Eads is the adsorption energy of the adjacent site; d1,d2 and d3 are the distance between the Al atom and the adjacent three Si atoms,h is the distance between the Al atom and the adjacent Si atom in the vertical direction.
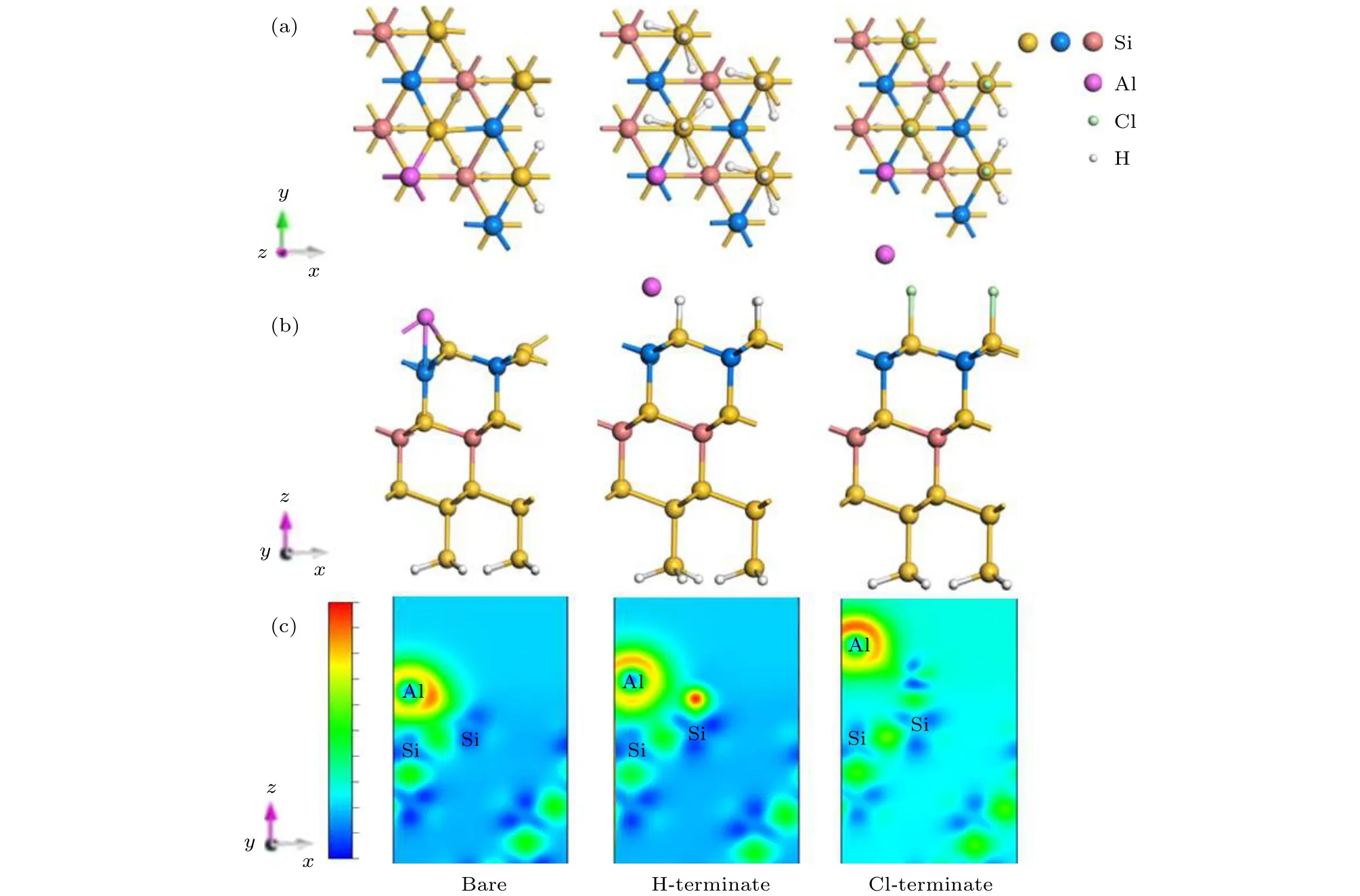
图6 Al原子吸附在清洁、H钝化、Cl钝化Si(111)表面的(a)俯视图,(b)侧视图,(c)差分电荷密度图Fig.6.Al atom adsorbed on clean,H-terminate,Cl-terminate Si(111) surface:(a) Top view; (b) side view; (c) differential charge image.
3.4 Al原子在Si(111)表面的扩散
如图7(a)所示,将Al原子吸附在最稳定的T4位点作为初始状态,吸附在H3位点作为最终状态,做NEB扩散研究.在清洁的Si(111)表面,Al原子吸附在T4位点时,与位于三个T1位点的Si原子成键,Al-Si键长为2.55 Å (表2),此时Al原子被3个Al-Si键牢牢固定,因而在T4位点的Al原子的吸附能最大,状态最稳定; 随着Al原子扩散迁移,Al原子与反方向T1位点的键出现了断裂; 当Al原子迁移到鞍点位置时,Al原子与邻近的两个T1位点的Si原子成键,键长为2.56 Å,比初始状态的Al-Si键短了0.06 Å,此时扩散的能垒最大为0.65 eV (表3),Al原子最不容易扩散迁移.Al原子在扩散路径上的竖直高度基本保持不变,其中吸附在H3位点的Al原子只比吸附在初始位置T4位点的Al原子在竖直高度上低0.003 Å.通过图6(a)和图6(b) (中图,右图)可见,在H钝化和Cl钝化Si(111)表面,由于H原子或者Cl原子饱和了Si表面的悬挂键,Al原子未与表面Si原子成键,扩散能垒明显降低.从表3可知,Al原子在清洁的、H钝化、Cl钝化Si(111)表面扩散的路径均为从T4位点沿直线到H3位点,并非绕顶部Si原子.Al原子在H钝化Si(111)表面扩散路径上的竖直高度呈下降趋势,吸附在最低处H3位点的Al原子比吸附在初始位置时低0.007 Å.Al原子在Cl钝化Si(111)表面扩散路径上的竖直高度出现先上升后下降的趋势,最高点位于鞍点,比吸附在初始位置时高0.004 Å.从T4位点直接到相邻T4′[15],扩散能垒分别为0.65,0.05,0.14 eV.因此,H原子或者Cl原子处理Si(111)表面均有效地降低了Al原子的扩散能垒,使Al原子在Si表面的扩散变得更加容易.
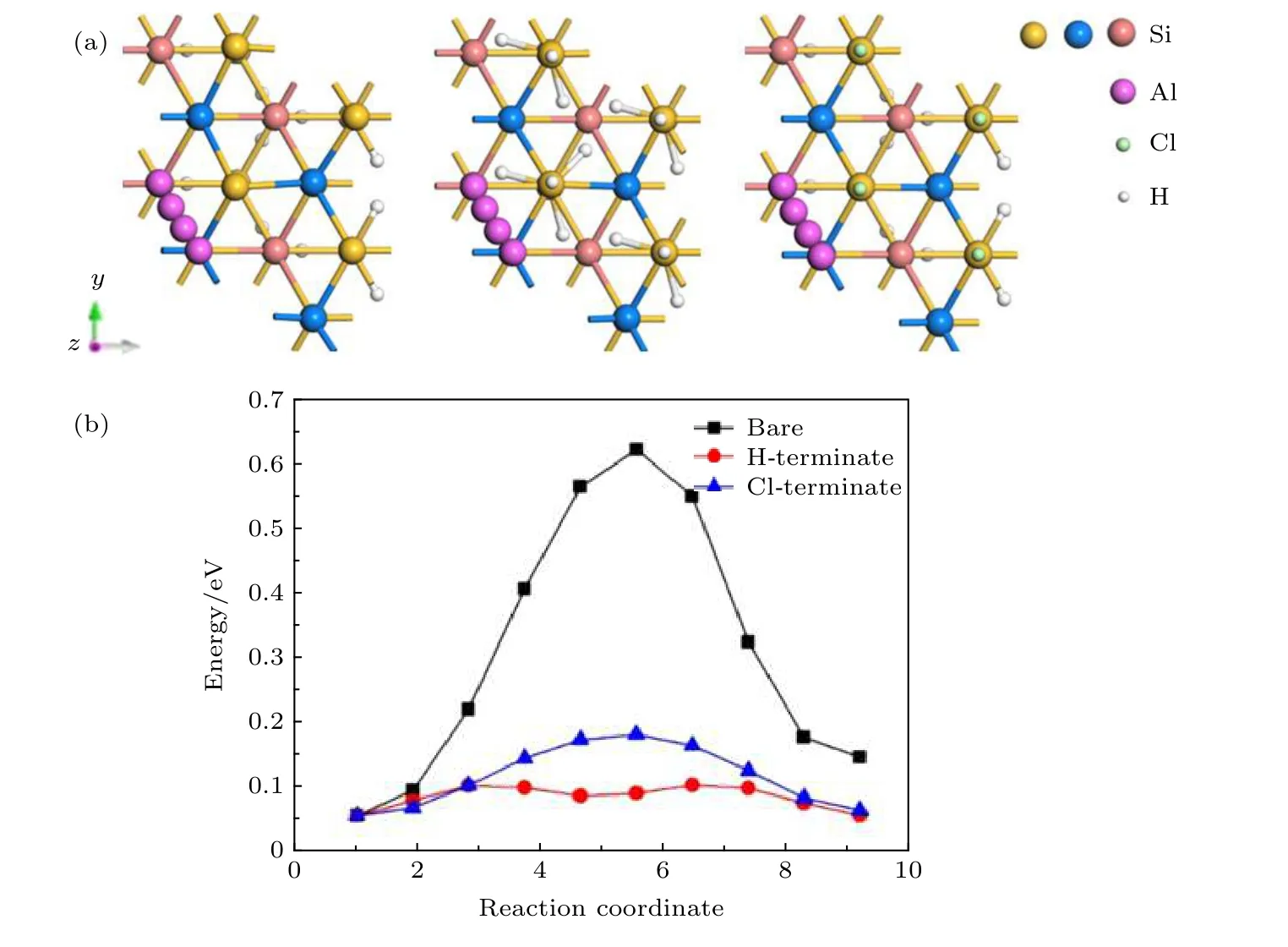
图7 Al原子在清洁、H钝化、Cl钝化Si(111)表面 (a)扩散路径; (b)扩散能垒Fig.7.Al atoms on clean,H-terminate,and Cl-terminate Si(111) surfaces:(a) Diffusion paths; (b) diffusion barriers.
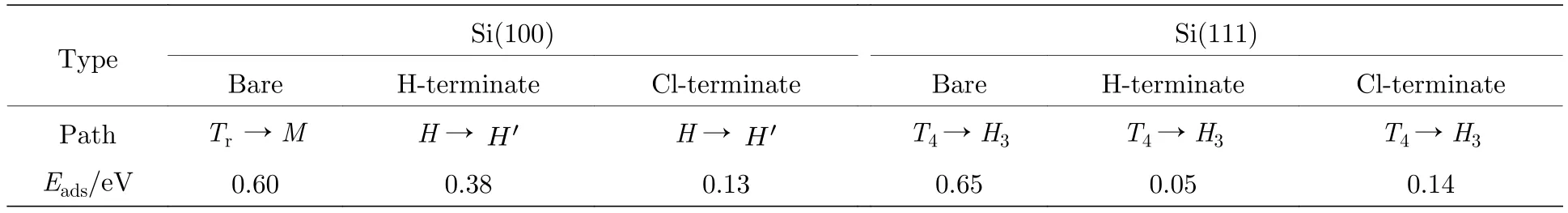
表3 Al原子吸附在清洁、H钝化、Cl钝化Si(100)和Si(111)表面的扩散路径和扩散能垒Table 3.Diffusion path and diffusion energy of Al atom adsorbed on clean,H-terminate,Cl-terminate Si(100) and Si(111)surfaces.
4 总 结
基于密度泛函理论的第一性原理计算,我们从原子尺度研究了Si表面在H或Cl钝化处理后,对单个Al原子在其表面的吸附和扩散行为的影响.通过NEB过渡态搜索的计算,获得了最优化的Al原子的迁移路径和不同钝化表面的扩散能垒.计算结果表明:1)在不同方式处理的Si(100)表面,Al原子吸附的最稳定位置不同、扩散路径也不同,清洁表面上,Al易于吸附在沟槽中的Tr位点,沿沟槽呈曲折状扩散; H钝化、Cl钝化的表面上,Al易于吸附在二聚体列顶部的H位置,在二聚体列顶部沿直线扩散; 2) Al原子在清洁、H钝化、Cl钝化Si(111)表面稳定吸附位点相同均为T4位点,扩散路径完全相同,均从Si(111)表面T4位点扩散到H3位点; 3)无论是Si(100)还是Si(111)表面,H钝化、Cl钝化处理均有效降低Al原子的扩散势垒.本研究结果有助于理解不同Si表面状态对外延生长的调控机制,尤其是为利用金属Al层作为缓冲层生长外延氮化物的工艺优化提供理论支撑.
