基于综合评分法优选猪苓最佳炮制方法*
2019-10-24李丽红刘丽婷刘亚男窦志英
李丽红,刘丽婷,刘亚男,毛 睿,王 飘,窦志英
(天津中医药大学,天津 301617)
猪苓为多孔菌科真菌猪苓[Polyporus umbellatus(Pers.)Fries]的干燥菌核[1],其菌核入药在中国已有2 000多年的历史,具有利水渗湿之功效[2],临床上主要用于治疗小便不利、水肿、泄泻等症。其分布较广,以云南产量最大、陕西所产质量为优[3]。猪苓的生长发育一般要经过4个阶段,分别是担孢子、菌丝体、菌核和子实体[4],在这期间猪苓主要依靠蜜环菌供给其生长所需要的营养物质[5]。猪苓的化学成分较多,其中多糖类成分是猪苓的重要活性成分,具有抗肿瘤、延缓衰老、保肝等作用[6-8],甾体类成分是主要的利尿成分,此外还有氨基酸类、维生素类及微量无机元素等[9]。
合理的炮制方法不仅可以保证药材的质量,还能降低成本,提高效率以及减少药材的损耗[10]。猪苓采收后均需要在产地净选去除杂质后,再采用水处理软化至内外湿度均匀一致[11],按照一定的规格切制成饮片,猪苓饮片可直接用于临床调配处方或作为供药制剂的原料。王天媛等[12]采用晒干、阴干、不同温度烘干共10种加工方法处理猪苓药材,结果发现不同加工方法处理的猪苓药材性状差异较小,有效成分含量存在显著性差异,40、50℃烘干处理时麦角甾醇和总多糖含量较高。鲁文静等[13]研究结果表明,50℃烘干法的多糖含量最高,阴干法多糖含量较低;自然晒干及70℃烘干所得麦角甾醇含量较高,而高温烘干得到的麦角甾醇含量较低。而到目前为止,对猪苓的浸润方法研究较少。本实验采用不同浸泡时间和不同干燥方法对猪苓药材进行加工处理,以猪苓中麦角甾醇、麦角甾酮、多糖和水溶性浸出物为评价指标,采用综合评分法优选猪苓的最佳炮制方法,为猪苓的炮制工艺及质量控制提供参考。
1 仪器与材料
1.1 仪器 LC-20A高效液相色谱仪(日本岛津公司);色谱柱为phenomenon-C18(250 mm×4.6 mm,5 μm);UV6100S型紫外分光光度仪(上海美普达仪器有限公司);KQ-300B超声波清洗仪(昆山市超声仪器有限公司);FA210电子分析天平(上海舜宇恒科学仪器有限公司);FW100高速万能粉碎机(天津市泰斯特仪器有限公司);HH-4数显恒温水浴锅(江苏金坛市科析仪器有限公司);XMT-921A远红外恒温干燥箱(天津市华北实验仪器有限公司);SHZ-D(Ⅲ)循环水式多用真空泵(巩义市英峪高科仪器厂)。
1.2 材料 麦角甾酮对照品(天津蓝天翔宇科技有限公司,批号X27J8L38809)、麦角甾醇对照品(上海生工生物有限公司,批号A600443-0005)、D-无水葡萄糖对照品(中国食品药品检定研究院,批号110833-201707);蒽酮(天津索罗门生物科技有限公司);95%乙醇(分析纯,天津渤化化学试剂有限公司);浓硫酸(天津市化学试剂供销公司);纯净水(杭州娃哈哈集团有限公司)。
猪苓药材来源于陕西省商洛市丹凤县,经天津中医药大学李天祥教授鉴定为多孔菌科真菌猪苓[Polyporus umbellatus(Pers.)Fries]的干燥菌核,样品粉碎过4号筛。
2 实验方法
2.1 麦角甾醇和麦角甾酮含量测定
2.1.1 色谱条件色谱柱为phenomenon-C18(250mm×4.6 mm,5 μm);流动相为甲醇-水(95∶5);检测波长麦角甾醇283 nm,麦角甾酮343 nm,体积流量1.0 mL/min;柱温:30℃。
2.1.2 对照品溶液制备 精密称取麦角甾醇和麦角甾酮对照品适量,加甲醇溶解,得浓度分别为0.400 mg/mL麦角甾醇、0.048 mg/mL麦角甾酮对照品溶液。
2.1.3 供试品溶液制备 取猪苓样品粉末(过4号筛)0.5 g,精密称定,置于100 mL具塞三角瓶中,加入甲醇10 mL,称质量,超声60 min,放至室温,加溶剂补足质量,混匀,滤过,用0.45 μm微孔滤膜滤过,取续滤液备用。
2.1.4 方法学考察 线性关系:精密移取麦角甾醇、麦角甾酮对照品溶液适量,用甲醇稀释一系列浓度的对照品溶液,麦角甾醇浓度分别为0.400、0.200、0.100、0.050 0、0.025 0、0.0125、0.0 0600、0.003 00 mg/mL,麦角甾酮浓度分别为 0.048 0、0.024 0、0.012 0、0.006 0、0.003 0、0.001 5、0.000 80、0.000 40 mg/mL,进样量20 μL,注入高效液相色谱仪,按上述色谱条件测定峰面积,以峰面积为纵坐标(Y),质量浓度为横坐标(X),绘制标准曲线,回归方程麦角甾醇为Y=34 283 478X+58 072,相关系数 r=0.999 3,线性范围:0.003~0.400 mg/mL。麦角甾酮为Y=44 843 619X+13 375,相关系数 r=0.999 1,线性范围:0.000 4~0.048 0 mg/mL。
精密度实验:取“2.1.2”项下混标溶液,在色谱条件“2.1.1”项下,连续进样6针,测定峰面积并计算RSD值。结果显示麦角甾醇和麦角甾酮RSD值分别为0.57%、1.04%,表明仪器精密度良好。
稳定性实验:取同一供试品溶液,按“2.1.1”项下色谱条件在 0、2、4、8、12、24 h 分别进样,记录峰面积并计算RSD值。结果麦角甾醇和麦角甾酮RSD值分别为1.54%,1.95%,表明供试品溶液在24 h内稳定性良好。
重复性实验:精密称取同一个样品6份,按“2.1.3”提取处理方法制备供试品溶液,在“2.1.1”项下色谱条件进行分析,测定峰面积,计算各对照品的质量分数,结果麦角甾醇和麦角甾酮RSD值分别为0.81%,0.69%,表明重复性良好。
加样回收率测定:精密称取9份已测定的猪苓粉末各约0.25g,分别加入麦角甾醇、麦角甾酮对照品溶液(相当于猪苓饮片中麦角甾醇、麦角甾酮的80%,100%,120%的含量),按“2.1.3”项下方法制备供试品溶液,按“2.1.1”项下色谱条件进行分析,测定峰面积,计算回收率及RSD值。结果麦角甾醇、麦角甾酮的加样回收率分别为99.93%、99.86%,RSD值分别为1.51%、1.78%。
2.2 多糖含量测定
2.2.1 硫酸-蒽酮试剂的配制及检测条件 取蒽酮0.2 g溶解于100 mL浓硫酸中,即得,当天配制使用。取1 mL多糖供试液至25 mL具塞试管中,冰水浴中缓慢滴入2.5 mL硫酸蒽酮,摇匀,置于沸水浴中加热10 min,取出,冰水浴冷却后,以蒸馏水做空白对照,626 nm下测得吸光度,并根据吸光度计算猪苓多糖含量。
2.2.2 对照品溶液的制备 取无水葡萄糖标准品(105℃干燥至恒重)100 mg,精密称定,置100 mL量瓶中,加蒸馏水定容至刻度,摇匀,即得1 mg/mL的葡萄糖对照品储备液。
2.2.3 供试品溶液的制备 取猪苓粉末约1.0 g,精密称定,置于250 mL锥形瓶中,向其加入80 mL 95%乙醇超声20 min,过滤,弃去滤液,回收滤渣,滤纸及滤渣挥干溶剂后放锥形瓶中,加水80 mL,超声提取20 min,过滤得滤液,转移定容至100 mL容量瓶中,取1 mL溶液稀释至10 mL容量瓶中即得多糖供试液。
2.2.4 方法学考察 标准曲线的绘制:精密吸取上述葡萄糖对照品储备液 0、0.1、0.2、0.4、0.6、0.8、1 mL置10 mL容量瓶中,加蒸馏水定容至刻度,分别取1 mL置10 mL具塞试管中,各加入硫酸蒽酮溶液2.5 mL,混匀,置沸水浴中加热10 min,冰水浴中冷却降温,另取蒸馏水1.0 mL同上平行操作,作为空白溶液,在626 nm下测定吸光度。以吸光度为纵坐标(Y),以葡萄糖溶液浓度为横坐标(X)进行线性回归,得回归方程Y=9.599 1X+0.069 3,r=0.998 4,线性范围 0~1.013 mg/mL。
精密度实验RSD值为0.290%,表明仪器精密度良好;重复性实验RSD值为1.83%,表明实验重复性良好;稳定性实验RSD值为1.98%,表明样品在2 h内稳定;加样回收率实验平均加样回收率为99.96%,RSD值为1.89%。
2.3 浸出物含量测定 水溶性浸出物测定:按2015年版《中华人民共和国药典》第四部附录2201水溶性浸出物测定方法(冷浸法)对样品进行测定,计算水溶性浸出物的量。
3 炮制工艺研究
3.1 优选最佳浸泡时间
3.1.1 最大吸水量考察 取相同规格的猪苓药材100 g,平行 3份,各加水 500 mL,浸泡 24 h,考察最大吸水量。结果24 h内最大吸水量为(130.00±2.00)mL。
3.1.2 吸水速率考察 取相同规格的猪苓药材100g,平行 3 份,各加水 500 mL,分别在浸泡 2、4、6、8、10、12、24 h时测定其吸水量,并计算吸水速率。结果见表1。
表1 不同浸泡时间吸水速率
吸水速率(mL/h)35.33±2.57 7.17±0.58 4.83±0.14 3.67±0.26 3.67±0.89 3.00±0.32 1.94±0.29浸泡时间(h) n 0~2 3 2~4 3 4~6 3 6~8 3 8~10 3 10~12 3 12~24 3
由表1可知,随着浸泡时间的延长,猪苓药材吸水速率逐渐减慢,0~2h吸水速率最快,为35.33mL/h,8 h前后吸水速率基本保持不变,说明猪苓药材在浸泡8 h基本达到饱和状态,且药材已达到润软可切的程度。
3.1.3 浸泡时间考察 取同一产地同一规格净制后的猪苓药材各100 g,分别加等量的水并用湿纱布覆盖于表面,分别浸泡 2、4、6、8、10、12、24 h,每组平行3份,切片,干燥。测定不同浸泡时间下猪苓中麦角甾醇、麦角甾酮、多糖和水溶性浸出物的含量,测定结果见表2。
3.1.4 采用综合评分法优选猪苓药材最佳浸泡时间 采用综合评分法来选择最佳浸泡时间,隶属度函数计算:F(x)=(x-x最小值)/(x最大值-x最小值);综合分数=多糖隶属度×50%+麦角甾醇隶属度×30%+麦角甾酮隶属度×10%+水溶性浸出物隶属度×10%。结果见表2。
由表2可知,猪苓药材在浸泡2~8 h时间段内,麦角甾醇和麦角甾酮含量均有所增加,浸泡8 h时二者含量均达到最大值。其中浸泡2 h与4 h、4 h与6 h麦角甾醇无显著性差异(P>0.05),浸泡8 h与浸泡时间<8 h各组之间均存在极显著性差异(P<0.01)。浸泡8 h之后,两者含量又有所降低。而多糖和水溶性浸出物含量随着浸泡时间的延长,都呈现先升高后下降的趋势,浸泡10 h达到最大值,其中,浸泡2~6 h之间各组均无显著性差异(P>0.05),浸泡8 h与浸泡时间<8 h各组之间有显著性差异。
由综合评分结果可知,浸泡8 h综合评分最高,其分值达到0.796,浸泡时间过短和过长综合评分值都较低,浸泡24 h综合评分分值仅为0.254,说明浸泡24 h有效成分损失较大。由此可得出猪苓药材不宜浸泡过长时间,最佳水浸泡时间为8 h。
3.2 干燥方法的优选 取同一批猪苓药材按上述优选的最佳浸泡时间浸泡8 h后,切片,分别采用烘干法(50℃、60℃、70℃、80℃烘干)、自然晾干法、冷冻干燥法、微波干燥法进行干燥,每组平行3份。
3.2.1 不同干燥方法对各指标含量的影响 测定不同干燥方法猪苓中麦角甾醇、麦角甾酮、多糖、水溶性浸出物的含量,研究不同干燥方法对其含量的影响。测定结果见表3。

表2 优选猪苓药材最佳浸泡时间的综合评分表
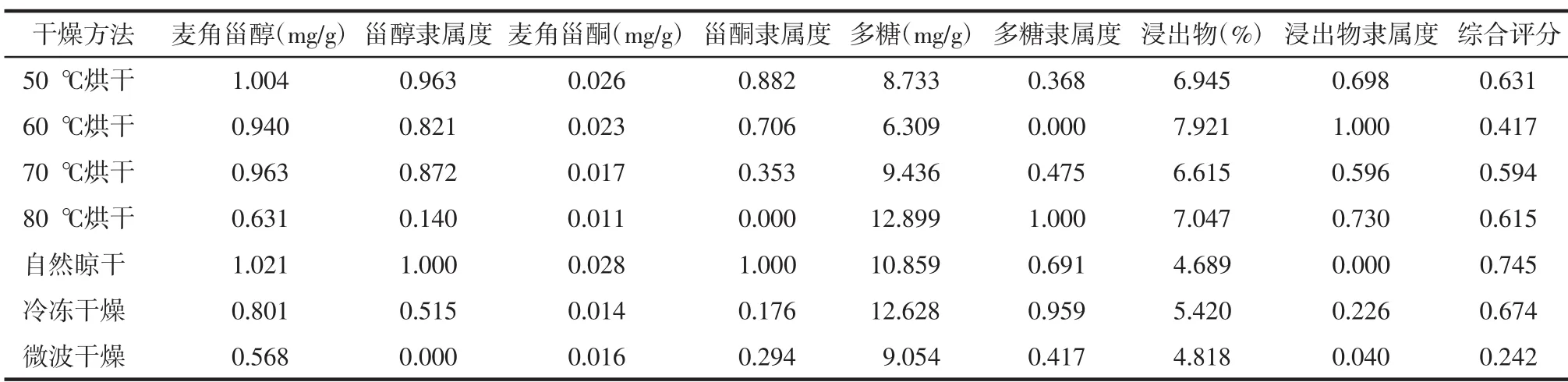
表3 优选猪苓最佳干燥方法的综合评分表
3.2.2 采用综合评分法优选猪苓最佳干燥方法 以麦角甾醇、麦角甾酮为指标,最佳干燥方法为自然晾干,其次是50℃烘干;以多糖为指标,最佳干燥方法为80℃烘干;以水溶性浸出物为指标,最佳干燥方法为60℃烘干。说明不同干燥方法对猪苓中的各成分含量有很大的影响,因此采用综合评分法优选最佳干燥方法。隶属度函数计算公式见“3.1.4”,计算结果见表3。
结果表明,不同干燥方法对各成分含量有显著影响,自然晾干下麦角甾醇、麦角甾酮含量最高,其次为50℃烘干。不同干燥方法中麦角甾醇含量大小顺序为:自然晾干>50℃烘干>70℃烘干>60℃烘干>冷冻干燥>80℃烘干>微波干燥,其中,50℃烘干麦角甾醇含量显著高于80℃烘干,而与自然晾干无显著性差异(P>0.05)。麦角甾酮含量大小顺序为:自然晾干>50℃烘干>60℃烘干>70℃烘干>微波干燥>冷冻干燥>80℃烘干,各组之间均有显著性差异(P<0.05)。多糖含量大小顺序为:80℃烘干>冷冻干燥>自然晾干>70℃烘干>微波干燥>50℃烘干>60℃烘干。其中,50℃烘干与60℃、70℃烘干无显著性差异,与80℃烘干有显著性差异(P<0.05)。浸出物含量大小顺序为:60℃烘干>80℃烘干>50℃烘干>70℃烘干>冷冻干燥>微波干燥>自然晾干。其中,60℃烘干与各组之间均有显著性差异。
由综合评分结果可知,干燥方法中自然晾干的综合评分最高,其分值达0.745,其次是冷冻干燥,其分值为0.674,50℃烘干,其分值为0.631,评分最低的为微波干燥,分值为0.242。由于自然晾干时间较长,且受天气影响;冷冻干燥成本较高,操作繁琐,且耗时长,不能广泛采用。所以,选择50℃烘干作为猪苓干燥加工方法较为合适,适用于大规模生产。由此表明,综合评分法是一种比较合理的评估药材质量的一种简单可靠的方法。
4 讨论
中药材的水处理是一个重要环节,掌握正确的水处理方法,可以提高药材有效成分的利用率[14-16]。但是目前仍存在着中药材浸润工艺不规范和不合理的操作,特别是将药材长时间浸泡在水中,会致使其有效成分大量流失,甚则发霉腐烂,严重影响药材饮片或中成药的内在质量甚或产生毒性物质。2015年版《中华人民共和国药典》规定,猪苓药材的炮制方法为洗净,浸泡,润透,切片,但对于具体的浸泡时间没有规定,各省市炮制规范中对猪苓药材的浸润方法也不尽相同,而且到目前为止,有关猪苓浸润及干燥工艺的研究较少,因此本研究考察了猪苓药材浸泡时间及干燥方法,通过综合评分法优选最佳浸泡时间及干燥方法。
结果表明,浸泡8 h综合评分分值最高,达到0.796,且药材软化程度较好,切出的饮片较美观,同时有效成分含量较高。干燥方法中,自然晾干法综合评分最高,达0.745,但对于大规模集中生产来说,这种方法既不能提高生产效率又不易达到药材的卫生要求,且受天气影响比较大。而烘干法时间短,且温度好控制,适合大规模生产。烘干法中50℃烘干综合评分值最大,因此综合考虑采用50℃烘干较为合适。该干燥方法的优选为猪苓的大规模生产及质量控制提供参考。
