人工耳蜗植入患者常见聋病相关基因突变与内耳畸形的相关性分析
2019-10-24赵晓云胡健仵倩刘贝贝陈迟边盼盼郭玉芬徐百成
赵晓云 胡健 仵倩 刘贝贝 陈迟 边盼盼 郭玉芬 徐百成
兰州大学第二医院耳鼻咽喉头颈外科(兰州730030)
Conflict of Interest:No conflict of interest was declared by the authors.儿童听力损失发生时间、持续时间和严重程度都不同程度地影响其言语、语言、心理、学习和社交等方面的发展,先天性双耳重度至极重度听力损失对这些的影响尤为明显。国外研究表明,先天性听力损失的发病率为1‰-3‰,其中重度至极重度听力损失的发生率约为1‰[1]。我国多地流行病学研究显示重度至极重度听力损失约占先天性听力损失的1∕3[2-4],目前国内外研究显示非综合征感音神经性听力损失与SLC26A4和GJB2基因突变最为相关。国内外对于先天性感音神经性听力损失的分子病因和形态发育的相关性开展了一些研究,部分先天性听力损失可通过影像学检查发现骨迷路异常。目前双侧重度至极重度感音神经性听力损失的主要有效治疗手段为人工耳蜗植入(Cochlear Implantation,CI),多数患者在接受CI手术后可以获得不同程度的听力言语康复效果。明确CI群体的分子病因学及与内耳畸形的关系,回顾性分析这些影响因素与CI术后的听觉言语康复效果,有助于进一步指导临床实践。本研究旨在研究听力损失发生与常见聋病相关基因突变的联系,并进一步分析基因突变和内耳畸形的内在联系,研究开展遗传咨询和人工耳蜗植入术前评估的科学途径。
1 研究对象与方法
1.1 研究对象
以2012年-2018年于兰州大学第二医院接受单侧CI手术并坚持系统的听力言语康复训练的250例患者为研究对象,所有患者手术过程顺利,术后无并发症,其中男性134例,女性116例。所有250例患者有119例在3岁内(包括3岁)完成CI手术,有114例在4-7岁接受CI手术。
1.2 研究方法
1.2.1 聋病相关基因突变检测
征得患者监护人同意后签订遗传性耳聋基因检测知情同意书,采集静脉后使用华大基因公司的磁珠提取纯化试剂盒提取外周血基因组DNA,设计特异性扩增引物后利用多重PCR技术扩增目的片段进行纯化,采用定制的基因片段捕获芯片(Roche NimbleGen,Madison,USA)与标记好的患者DNA库杂交,构建文库,进行质检,最后利用高通量测序仪Illumina HiSeq2500 Analyzers(Illumina,SanDiego,USA)测序,并使用常见聋病相关基因包括GJB2、SLC26A4、GJB3、mtDNA、OTOF等22个基因共159个位点耳聋信息分析流程进行分析。
1.2.2 听力学检查
患者均行听性脑干反应(ABR)、听觉稳态诱发电位(ASSR)、畸变产物耳声发射(DPOAE)及声导抗测试,同时根据年龄进行对应的行为测听检查。听力损失分级以行为测听为主,并结合客观听力检查进行评价,以0.5、1、2KHz平均听阈计算(WHO,1980):<25dBHL为正常听力,26-40dBHL为轻度听力损失,41-55dBHL为中度听力损失,56-70dBHL为中重度听力损失,71-90dBHL为重度听力损失,>91dBHL为极重度听力损失。本研究所有患者均表现为重度至极重度听力损失。
1.2.3 影像学检查
所有患者均行颞骨高分辨率CT(HRCT)、头颅MRI及内耳水成像检查。内耳畸形根据Sennaroglu2017标准进行分类,本研究中主要的内耳畸形表现为大前庭水管综合征(Large vestibular aqueduct syndrome,LVAS)、LVAS伴不完全分隔Ⅱ型(Incomplete partition typeⅡ,IP-Ⅱ)及单纯IP-Ⅱ,部分表现为共同腔畸形、内听道狭窄及半规管发育不良等。
1.2.4 统计学方法
使用SPSS 25.0软件进行统计分析,采用连续性校正卡方检验及Fisher确切概率法检验,P<0.05为差异具有统计学意义。
2 结果
2.1 人工耳蜗植入患者基因突变分布及突变频率
所有250例患者中发现已被公认的基因突变有 77例,突变频率为 30.80%(77∕250),主要为SLC26A4和GJB2基因突变。共检测到SLC26A4基因13种序列改变,其中SLC26A4双等位基因突变(纯合和复合杂合)15例,突变频率为6%(15∕250),有23例患者检出c.919-2A>G突变,是该基因检出率最高的突变形式,占SLC26A4基因突变患者的67.65%(23∕34),等位基因频率为5.2%(3例纯合突变,8例复合杂合突变,12例杂合突变)。共检测到GJB2基因7种致病突变,其中GJB2双等位基因突变23例,突变频率为9.2%(23∕250),最常见的是c.235delC和c.299-300delAT突变,等位基因频率分别为5.4%(7例纯合突变,10例复合杂合突变,3例杂合突变)和3.8%(1例纯合突变,13例复合杂合突变,4例杂合突变)。250例患者中6例检出mtDNA 12SrRNA突变,突变频率为2.4%(6∕250),5例为同质突变,突变位点为m.1555A>G和m.961T>C。2例患者检出GJB3基因突变,突变频率为0.8%(2∕250),均为c.538C>T杂合突变。还有3例复合基因突变,分别为1例GJB2和mtDNA 12SrRNA复合突变、2例SLC26A4和GJB2复合突变,所有患者基因突变情况见表1。
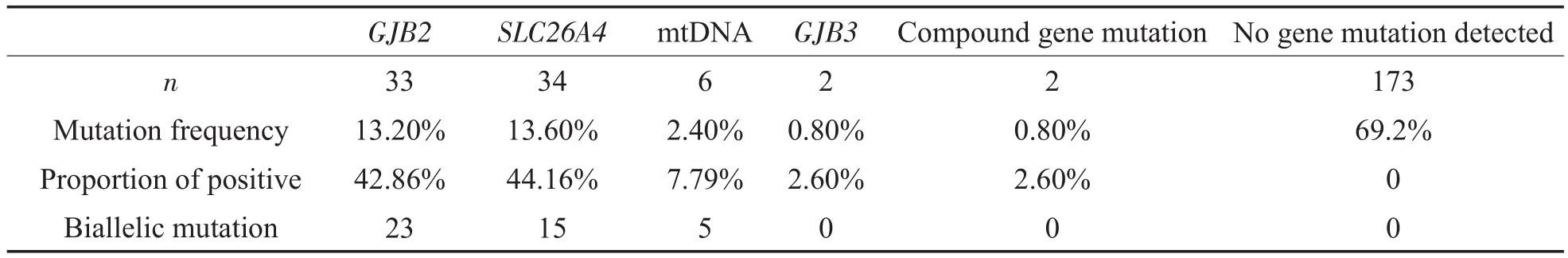
表1 250例患者的常见聋病相关基因突变检测结果Table 1 Detection results of common deafness-associated gene mutations in 250 patients
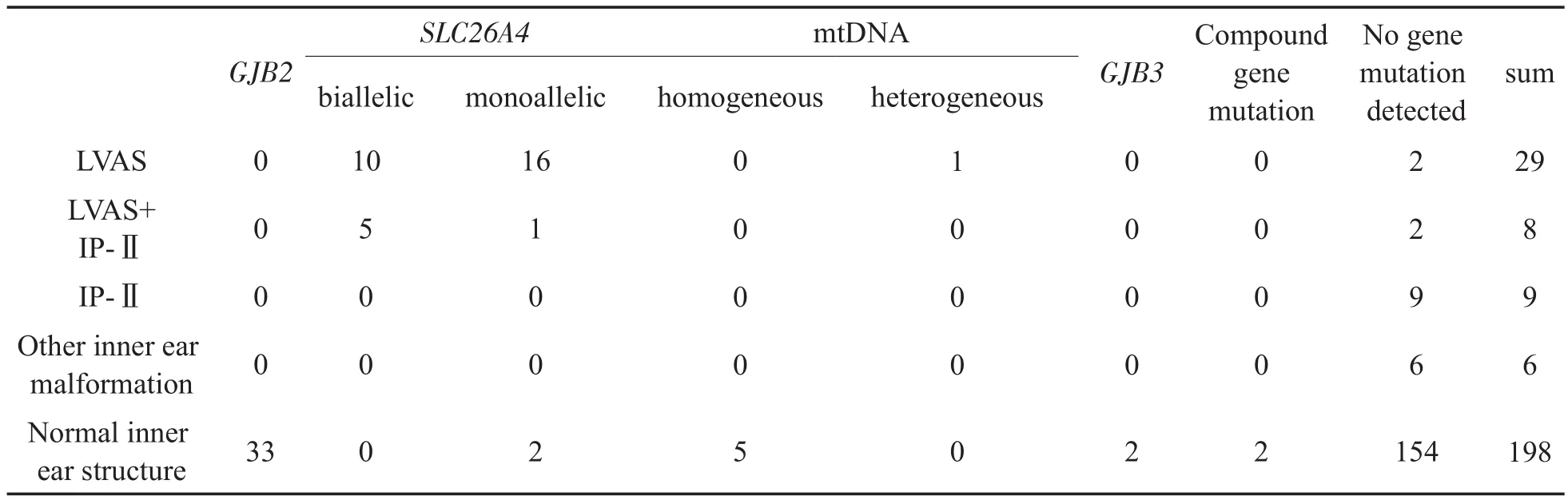
表2 250例人工耳蜗植入患者内耳畸形与基因突变的关系Table 2 The relationship between inner ear malformation and gene mutation in 250 patients with cochlear implantation
2.2 人工耳蜗植入患者内耳畸形分布情况
250例患者中,LVAS共29例,发生率为11.60%(29∕250),占内耳畸形的55.77%(29∕52);LVAS伴IP-Ⅱ8例,占内耳畸形的 15.38%(8∕52);单纯IP-Ⅱ9例,占内耳畸形的17.31%(9∕52)。其他内耳畸形包括共同腔畸形、内听道狭窄及半规管发育不良或未发育共6例;内耳结构正常198例,占该群体中的79.2%(198∕250),不同内耳畸形及分布见表2。
2.3 基因突变与内耳畸形的关系
在15例SLC26A4双等位基因突变患者中,2∕3为LVAS,1∕3为LVAS伴IP-Ⅱ;在19例SLC26A4单等位基因突变患者中,16例为LVAS,1例为LVAS伴IP-Ⅱ,2例内耳结构正常。而在所有的LVAS和LVAS伴IP-Ⅱ患者中,SLC26A4双等位基因突变占40.54%(15∕37),SLC26A4单等位基因突变占到了45.95%(17∕37),有1例携带mtDNA异质突变,4例未检测到常见聋病基因突变。对LVAS和LVAS伴IP-Ⅱ与内耳结构正常两组进行分析(见表3),统计学结果表明LVAS与内耳结构正常的SLC26A4基因突变携带率存在统计学差异(P<0.001),LVAS伴IP-Ⅱ与内耳结构正常的SLC26A4基因突变携带率存在统计学差异(P<0.001)。对单纯LVAS与LVAS伴IP-Ⅱ两组进行分析(见表4),统计学结果表明两组间SLC26A4基因突变结果的差异没有统计学意义(P=0.059)。在所有的GJB2基因突变患者中,未发现任何内耳畸形。在未检测到常见聋病基因突变的173例患者中,有2例LVAS,2例LVAS伴IP-Ⅱ,9例单纯IP-Ⅱ,6例其他内耳畸形(包括共同腔畸形、内听道狭窄及半规管发育不良或未发育),其余154例内耳结构正常。
3 讨论
有超过1∕3的听力损失与SLC26A4基因突变或GJB2基因突变相关[5]。有研究[6]显示SLC26A4基因突变相关听力损失程度以重度、极重度为主,GJB2基因突变相关听力损失程度以极重度为主。部分功能异常与器官组织的宏观和微观形态学差异有关,针对聋病的研究表明,约80%的先天性感音神经性听力损失与内耳膜性或者细胞水平的异常有关,这类疾病通过现有影像学检查无法诊断,而另外的约20%听力损失是由可通过影像学检查发现的各种骨迷路异常引起的,这一类患者常表现为重度及极重度听力损失[7]。目前国内外针对CI人群开展的基因突变研究样本均较小,开展更广范围的类似研究,可以明确CI人群的分子病因,并尝试阐述该群体内耳发育异常的病因与机制,为后续的CI疗效等研究提供依据。
本研究CI人群中常见聋病相关基因突变检出率 为 30.80%(77∕250),其 中 检 出 率 最 高 的 是SLC26A4基因突变(44.16%,34∕77)和GJB2基因突变(42.80%,33∕77),热点突变分别是c.919-2A>G和c.235delC、c.299-300delAT。我们的研究和国内外针对中国人群的研究结果一致:SLC26A4和GJB2基因突变是导致重度至极重度听力损失最常见的分子学病因,具有种族特异性的中国人的基因突变图谱SLC26A4为c.919-2A>G、GJB2为c.235delC。本研究中有一部分为单杂合突变,一部分未检测出常见聋病相关基因突变,其原因可能和我们的基因检测策略中未纳入的基因突变位点有关,也可能和其他基因突变、影响表型的修饰基因存在、表观遗传学的影响以及环境因素等有关。明确该群体听力损失病因及基因突变类型,可以为未来精准医学模式的推广应用奠定基础。

表3 LVAS及LVAS+IP-Ⅱ组和内耳结构正常组与SLC26A4基因突变的关系Table 3 The relationship between SLC26A4 gene and LVAS group and normal inner ear malformation

表4 单纯LVAS组和LVAS伴IP-Ⅱ组与SLC26A4基因突变的关系Table 4 The relationship between SLC26A4 gene and LVAS and LVAS with IP-Ⅱ
针对CI患者开展聋病相关基因诊断,首要的目的是可以在术前对手术效果进行较精准的评估。根据目前对GJB2基因突变致聋机制的研究,不管是经典的K+中毒学说还是因Cx26缺陷导致耳蜗发育障碍引起听力损失的机制[8,9]都认为病变部位在耳蜗。SLC26A4基因突变主要导致耳蜗功能障碍和内耳发育异常[10]。线粒体基因突变主要与氨基糖苷类抗生素药物性聋有关,其会使线粒体呼吸链功能受影响,引起毛细胞萎缩、死亡[11],且药物易感性聋较药物中毒性聋对氨基糖苷类抗生素更加敏感。这三种最常见的聋病相关基因突变主要影响耳蜗及其内部的微结构,SLC26A4基因突变可能影响球囊和前庭,但都没有对蜗后突触、听神经和听觉高级中枢产生明显的影响,所以理论上认为作为替代疗法的人工耳蜗植入术在SLC26A4、GJB2及线粒体基因突变患者应该获得良好的效果。我们的术后听力言语康复效果研究中也印证了这个观点,与国内外研究相符[12-14]。
基于手术安全和术后效果考虑,在早期的术前筛查阶段,严重的内耳畸形和明显影响手术和效果的畸形未能进入本研究的范围。本研究中内耳畸形的检出率为20.80%(52∕250),其中主要为LVAS(55.77%,29∕52)、LVAS伴IP-Ⅱ(15.38%,8∕52)及单纯 IP-Ⅱ(17.31%,9∕52)。我们的研究表明,LVAS和IP-Ⅱ等内耳畸形也是CI手术的适应症,对此类患者进行CI手术多数可以获得良好的听力言语康复效果。本研究显示在所有的有LVAS的患者中SLC26A4基因突变占86.49%(32∕37),其中双等位基因突变占40.54%,而在198例内耳结构正常者中只有2例为SLC26A4单基因突变,未检测到双等位基因突变,对LVAS和LVAS伴IP-Ⅱ与内耳结构正常两组的SLC26A4基因突变结果进行分析,发现均有明显统计学差异(P值均小于0.001),可见LVAS和LVAS伴IP-Ⅱ与SLC26A4基因突变存在明确的相关性。此外,有17例(45.95%,17∕37)LVAS患者检测出SLC26A4单等位基因突变,1例mtDNA突变,2例未检测出常见聋病相关基因突变,由于SLC26A4基因突变为常染色体隐性遗传,一般来说双等位基因突变(纯合或复合杂合突变)才会表现出其致病性。LVAS的发生除了受到SLC26A4基因突变的作用外,可能也受到其他因素的影响。本次研究所筛查SLC26A4基因突变共60个位点,可能因为部分突变类型未能进入筛查范围,但其他一些因素也需要考虑:1.SLC26A4基因大片段缺失、内含子突变及调控序列突变等[15];2.LVAS与其他基因突变相关,内向整流钾通道KCNJ10和SLC26A4双基因调控模式[16]、转录因子基因FOXI1调控SLC26A4基因的表达[17];3.存在影响听力表型的修饰基因;4.SLC26A4基因在常染色体隐性遗传的同时可能具有部分显性遗传特性;5.环境因素导致LVAS。本研究表明9例单纯IP-Ⅱ未检测到常见聋病基因突变,可见单纯IP-Ⅱ的发生与SLC26A4基因突变可能没有明确关系。我们将单纯LVAS和LVAS伴IP-Ⅱ的SLC26A4基因突变结果进行了分析,发现两组的基因突变结果差异无统计学意义,这说明二者可能具有相同或相似的分子病因,另外我们也可以通过进一步分析伴和不伴IP-Ⅱ的LVAS患者的病理生理及临床表型(听力损失的程度、频率和性质以及CI术后听觉言语康复疗效)来阐释二者的关系。本研究中发现部分仅存在耳蜗中转和顶转融合而不伴发LVAS的患者,推测这些患者可能的病理生理为:1.单纯IP-Ⅱ的发生与LVAS无关,它是胚胎发育期间某种因素导致的发育停止,该种情况其重度至极重度听力损失发生机制可能与膜迷路的发育异常有关,也可能有其他导致听力损失的病因存在;2.内淋巴囊和内淋巴管有扩大,但无CT上可显示的前庭水管的扩大,该种情况听力损失发生的机制可能与伴有LVAS的机制相同,与内淋巴囊和内淋巴管的扩大有关。LVAS因其发生时间和严重性不同,畸形可能仅仅表现为LVAS或者因压力传入内耳导致不同程度蜗轴缺失的发生,其中之一表现为IP-Ⅱ。朱庆文等[18]的研究显示经典Mondini畸形与SLC26A4基因突变密切相关,而不伴LVAS的内耳畸形即使患者的耳蜗畸形程度与Mondini畸形相同或相近,但与SLC26A4基因突变关系并不大。
本研究中33例GJB2基因突变所致听力损失中未发现内耳畸形,显示GJB2基因突变可能与各种类型的骨迷路畸形无关,这与目前国内外研究结果基本相符[19-21]。从目前对GJB2基因突变导致听力损失的机制研究[9]来看,GJB2基因突变导致缝隙连接蛋白26(Cx26)的缺陷,影响miRNA在细胞间的交流及表达,miRNA会影响大量的信使RNA和沉默基因,从而影响多种基因的表达和调节,进而对耳蜗发育产生影响。但耳蜗发育是一个复杂的过程,涉及众多基因表达和调节的协调。到目前为止,Cx26缺陷导致miRNA细胞间交流紊乱是如何影响基因表达及调节来影响耳蜗发育的详细机制仍不清楚。也有研究认为[22]GJB2基因突变对耳蜗支持细胞(如柱细胞)发育的影响也是听力损失产生的一个潜在因素。其他常见内耳和颞骨畸形如半规管畸形、共同腔畸形、内听道狭窄等可能和常见的聋病相关基因如GJB3、mtDNA等基因突变无关,可能和其他遗传因素、胚胎期前8周有致畸剂暴露史、病毒感染等有关。
250例CI患者中,有154例患者(61.60%,154∕250)内耳结构正常,且未检测到常见聋病相关基因突变,考虑一部分为非常见聋病基因突变所致,Liu等[23]报道有24.4%的CI患者检测出非常见聋病基因突变:MYO15A,TMC1,MYH14,MYO13,ACTG1,COL11A2,DSPP,GRHL2和WFS1。除此之外,环境因素也是导致先天性听力损失的重要因素,如如低出生体重、早产、出生窒息、宫内感染、新生儿高胆红素血症、妊娠期不当使用特殊药物、脑膜炎、巨细胞病毒及风疹病毒感染等所致。有学者[24]用影像学、遗传学及儿科评估的阶梯式诊断方法对先天性听力损失患儿的病因进行了分析,发现仍有一部分(25%-45%)患者听力损失的病因不能明确,例如新生儿重症监护室住院时间>5天或者接受过度体膜氧合、辅助通气、化疗等均有可能影响听力,还有神经组织退行性病变、感觉运动神经病等也是听力损失发生的危险因素[25]。
开展CI患者的常见聋病相关基因诊断研究,可以揭示重度至极重度的CI人群听力损失与GJB2和SLC26A4基因突变的相关性,明确SLC26A4基因突变是LVAS的主要分子病因、LVAS与LVAS伴IP-Ⅱ存在明显相关性以及单纯IP-Ⅱ与SLC26A4基因突变无关、GJB2基因突变并非骨迷路发育异常的分子病因。针对CI患者开展分子生物学研究,通过改进遗传学筛查策略、并详细分析病史及影像学资料以明确CI人群的病因,并进一步探索不同分子病因和内耳畸形导致的重度及以上听力损失与CI术后疗效的相关性,必将为探索开展遗传咨询和人工耳蜗植入术前评估的科学途径提供依据。
