人血清结合珠蛋白的生物信息学分析
2019-10-23李秀荣任怡然高瀛岱
李秀荣 孙 雪 任怡然 高瀛岱*
血清结合珠蛋白(haptoglobin,Hp)又称触角蛋白,是一种酸性蛋白,Hp基因表达最初在肝脏中被发现,后又陆续发现在心脏、脾脏等表达,是广泛存在于人类和哺乳类动物的血清和体液中的α2糖蛋白[1]。Hp具有遗传多态性,是重要的血清遗传标志物,位于常染色体16q22,具有两个等位基因Hp1和Hp2,呈孟德尔共显性遗传,具有3种基本基因型Hp1-1、Hp2-1和Hp2-2[2]。Hp基因表型的不同源于自然选择和遗传漂变[3]。特异的表型与疾病类型有关,Hp1-1与易感性感染疾病相关性较大(如白血病),在白血病相关研究中发现,Hp1-1纯合子患白血病的概率要高于其他型,Hp1-1属于易感基因[4]。Hp2-1与动脉粥样硬化和自身免疫病相关性较大,Hp2-2与血管闭塞性疾病有关[5]。Hp主要作用是结合游离血红蛋白(heamoglobin,Hb),形成Hp-Hb复合物,Hb中释放的游离铁和亚铁血红素可参与氧化反应导致组织损伤,而Hp与Hb结合后通过巨噬细胞清道夫受体CD163介导的内吞作用进行降解和代谢[6]。Hp-Hb复合物无法通过肾小球的滤过,因而防止了血红蛋白对肾脏的损伤[7]。CD163又称M130,属于富含半胱氨酸的清道夫受体(scavenger receptor cysteine-rich,SRCR)家族成员之一,是一种从血浆清除Hp-Hb复合物的高表达的巨噬细胞膜蛋白受体,受IL-6、IL-10和糖皮质激素的调控[8-10]。Hp在临床上主要作为溶血性疾病诊断的标志物。
本研究应用生物信息学的方法对人血清Hp的结构、理化性质等进行预测和分析,为在溶血性疾病及其他肿瘤诊断和治疗等方面的研究提供理论依据和基础。
1 材料与方法
1.1 材料
在Uniprot在线数据库以“Haptoglobin”为关键词,搜索Hp的蛋白序列信息,并下载蛋白序列(P00738)。
1.2 方法
应用在线数据库和在线分析软件,对Hp氨基酸序列进行多项生物信息学的分析预测。
2 结果与分析
2.1 人血清Hp的理化性质分析
利用PlatParam在线蛋白分析软件对人血清Hp的氨基酸个数、蛋白分子量、等电点、总原子数、半衰期、蛋白不稳定系数以及总平均亲水性等理化性质进行预测分析,分析结果显示,人血清Hp氨基酸个数为406个;蛋白相对分子量为45205.31;总原子数为6308;等电点为6.13,表明Hp为酸性蛋白,半衰期为30 h;蛋白不稳定系数32.94,表明为稳定蛋白;总平均亲水性-0.421,为负值,表明具有一定亲水性。
2.2 人血清Hp跨膜区域分析
利用在线跨膜区域分析工具TMHMM软件,分析人血清Hp是否具有跨膜区域。分析结果显示,人血清Hp不具有跨膜区域,为胞内蛋白,而非跨膜蛋白,见图1。
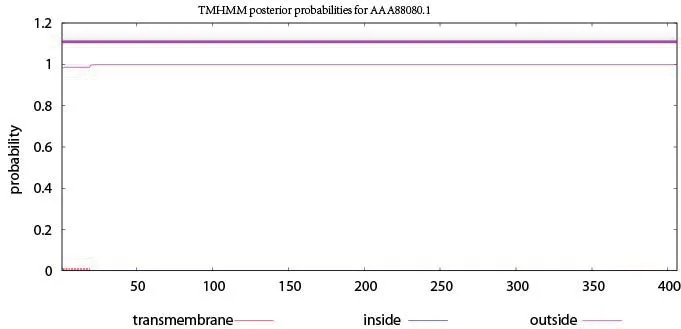
图1 人血清Hp跨膜区域预测分析
2.3 人血清Hp核定位序列预测分析
使用分析工具cNLS-Mapper对人血清Hp进行核定位序列分析,其结果显示,此蛋白有一段二元氨基酸序列 FDKSCAVAEYGVYVKVTSIQDWVQKTIAE,位于第377~406位,预测评分值(cut-off score)为2.4。根据文献[11]报道,当cut-off值为8~10时,表明蛋白质专一的定位于细胞核;当cut-off值为7或者8时,部分定位于细胞核;当cut-off值为3~5时,定位于核内和胞浆内;当cut-off值为1~2时,表明定位于胞浆内,不具有核定位序列。因此根据分析表明此蛋白不具有核定位序列,为胞浆蛋白,见图2。

图2 人血清Hp NLS预测分析
2.4 人血清Hp亲疏水性预测分析
通过ProtScale软件在线分析人血清Hp的亲疏水性,其结果显示:①最强疏水性位点为第8位氨基酸,异亮氨酸(Ile,I)得分为3.011;②最强的亲水性位点位于第73位氨基酸,谷氨酰胺(Gln,Q),得分为-2.456。预测结果显示,亲水性氨基酸比疏水性氨基酸多,此蛋白为亲水性蛋白,蛋白理化性质分析总平均亲水性(grand average of hydropathicity,GRAVY)为-0.421,为亲水性蛋白,与此分析结果一致,见图3。
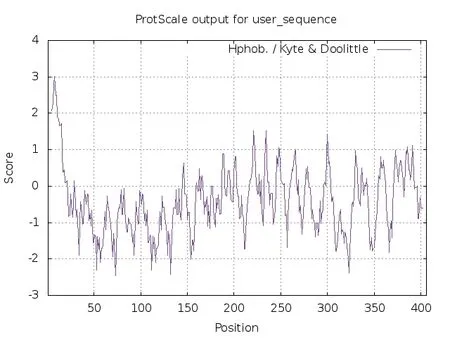
图3 人血清Hp亲疏水性预测分析
2.5 人血清Hp信号肽预测分析
通过SignalP 4.1 Sever在线分析软件,分析人血清Hp是否具有信号肽,cut-off值设置为0.5,分析结果显示:C、Y及S得分分别为0.692、0.807和0.971,分别位于第19、19和13位氨基酸上,Mean-S值为0.941,D值为0.879,D-cut-off为0.45,显示人血清Hp具有信号肽序列,见图4。

图4 人血清Hp信号肽序列预测分析

图5 人血清Hp二级结构预测
2.6 人血清Hp二级结构预测分析
通过Jpred 4在线分析软件,分析人血清Hp二级结构。输入人血清Hp氨基酸序列,发现蛋白结构数据库(protein Databank,PDB)内存在已知结构,可以继续预测分析,其结果显示:人血清Hp有16个β折叠结构,2个α螺旋结构,β折叠结构远远多于α螺旋结构,见图5。此结果只显示α螺旋和β折叠结构,其他信息用SOPMA蛋白结构分析软件进行分析比较。
用SOPMA在线分析软件对蛋白二级结构进行具体预测分析,其结果显示:人血清Hp氨基酸序列由14.29%的α螺旋、6.4%的β转角、24.38%的延伸链和54.93%的无规则卷曲构成,无规则卷曲为人血清Hp的主要折叠形式,α螺旋多于β转角,见图6。
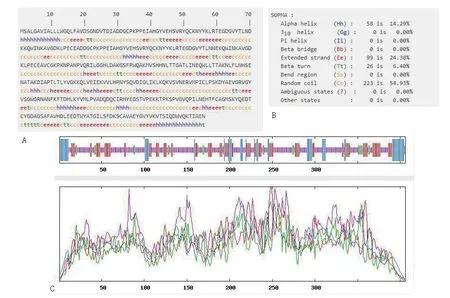
图6 人血清Hp二级结构预测分析
2.7 人血清Hp三级结构预测分析
Swiss-model软件是基于同源建模的方法,实现对一段未知序列的三级结构的预测。高级结构与蛋白功能相关性较大,研究蛋白的功能必须要掌握蛋白的高级结构特征。通过Swiss-model在线分析软件,对人血清Hp进行三级结构预测和分析,其结果显示得到3个预测结构图,见图7。
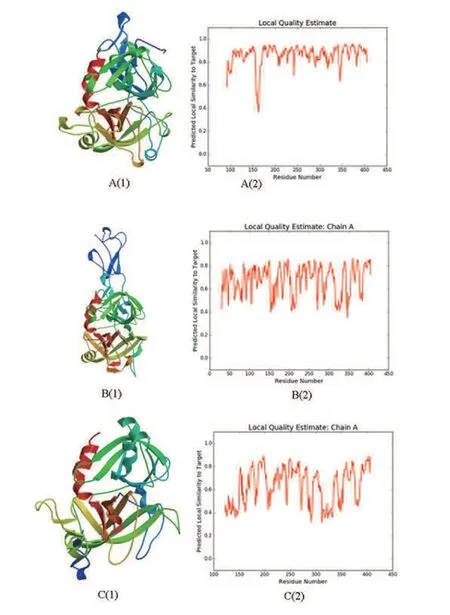
图7 人血清Hp三级结构预测分析
图7中A图预测结构与模板序列相似度为81.13%,相似性波形图值在0.73左右,覆盖度0.78;B图预测结构与模板序列相似度为32.96%,相似性波形图值在0.64左右,覆盖度0.88;C图预测结构与模板序列相似度为29.13%,相似性波形图值在0.45左右,覆盖度0.63。根据相似度、波形图及覆盖度判断模型A的预测结构可靠性更高。
为进一步验证模型A的可靠性,使用在线分析软件The Structure Analysis and Verification Server进行拉曼图谱分析,其结果显示:经预测所得的蛋白模型A除一个甘氨酸(符号标识:▲)外其余所有氨基酸残基都位于可接受区域内(颜色越深,接受度越高),第316位丝氨酸(Ser)和第227位赖氨酸(Lys)位于普通接受区,其余氨基酸都位于较高的接受区。结果表明所有氨基酸间能形成合理的二面角,使蛋白结构比较稳定,由Swiss-model软件基于同源建模的方法得到的人血清Hp三级结构的预测模型稳定可靠,见图8。
2.8 人血清Hp相互作用蛋白预测分析
通过String在线分析软件,对与人血清Hp相互作用的蛋白进行预测分析,其结果显示,有11个相互作用蛋白,并对这11个相互作用蛋白进行评分(见表1),预测了11种蛋白相互作用的网络图,见图9。
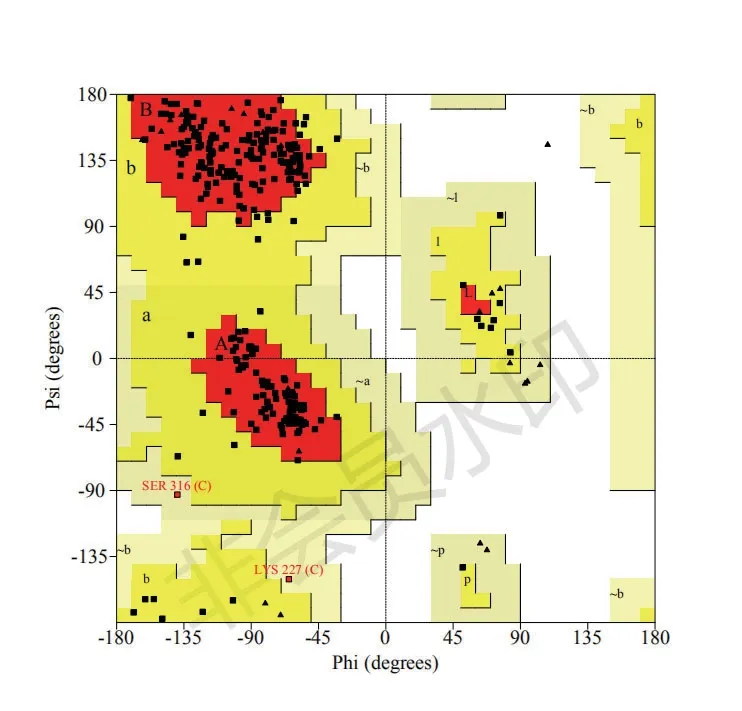
图8 人血清Hp预测三级结构模型A的拉曼图分析
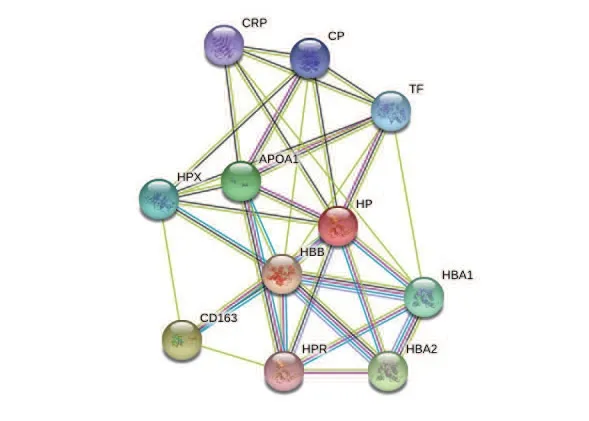
图9 与人血清Hp互相作用的蛋白预测分析
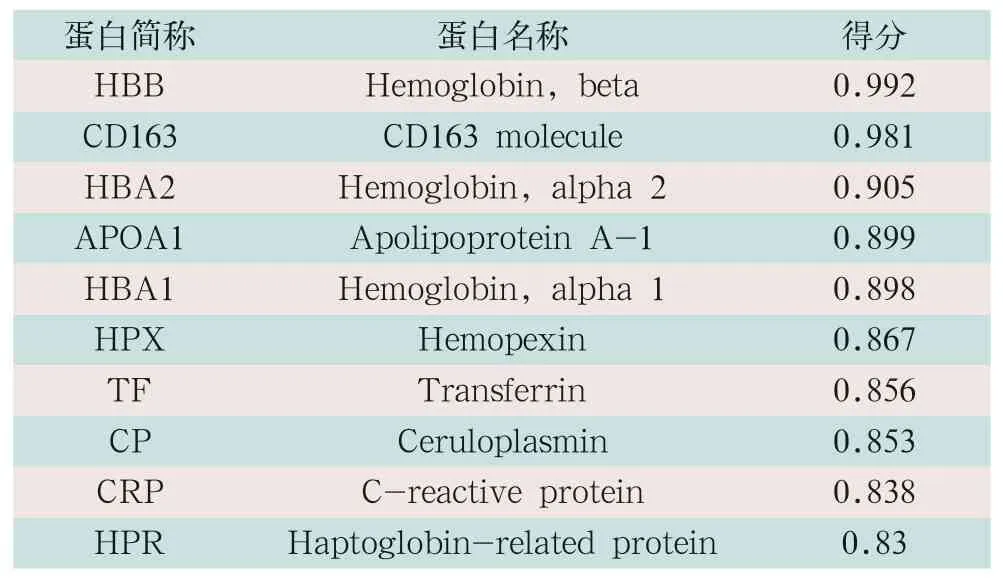
表1 与人结合蛋白互相作用的蛋白
Hp蛋白与Hb结合后,形成Hp-Hb复合物,通过CD163介导的内吞作用进行代谢,防止游离的血红蛋白对肾脏的损伤作用,与CPR互相作用,在炎症反应中发挥作用。APOA1为载脂蛋白A的一种,是卵磷脂胆固醇酰基转移酶的活化剂,该酶可将胆固醇转化为胆固醇酯,促进胆固醇的运输和调节HDL代谢,防止动脉硬化性心血管疾病的发生。人血清Hp蛋白还具有抗菌、抗氧化的作用。
2.9 人血清Hp功能注释分析
通过Gene Ontology在线分析软件对Hp进行功能(gene ontology,GO)注释,从生物学过程上来看,Hp主要参与蛋白质的水解作用,与Hb互相结合形成Hp-Hb复合物;参与受体介导的胞吞作用,Hp-Hb复合物通过巨噬细胞清道夫受体CD163介导的内吞作用进行降解、代谢;参与急性炎症反应;具有细胞死亡的正向调节作用;具有抗菌作用等。从细胞组分而言,Hp为胞浆蛋白,可与Hb形成Hp-Hb复合物。从分子功能而言,Hp具有蛋白结合作用;抗氧化活性;血红蛋白结合作用等。GO注释结果与蛋白互作结构一致,见表2。

表2 人血清Hp的 GO注释分析结果
3 讨论
人血清Hp是一种亲水性酸性蛋白,属于急性时相蛋白家族成员,与Hb结合形成Hp-Hb复合物,Hb产生游离铁和亚铁血红素可参与氧化反应,导致机体产生损伤,Hp-Hb复合物的产生防止Hb对机体的损伤作用。在血管内溶血性疾病,如溶血性贫血、阵发性睡眠性血红蛋白尿症(paroxysmal nocturnal hemoglobinuria,PNH)、遗传性球形红细胞增多症(hereditary spherocytosis,HS);血清学指标显著下降,被认定为一项临床诊断指标。Hp参与炎性反应,具有抗菌、抗氧化作用,促进血管生成作用,与冠心病、糖尿病、高血压、血液疾病、自身免疫病及恶性肿瘤都有相关性,还具有抗氧化活性、抗菌作用、抑制一氧化碳、血管生成、调节机体免疫功能等作用。Hp是一种急性期蛋白,在应激状态时,血清浓度升高;在发生溶血时血清浓度降低。当发生血管内溶血时,如溶血性贫血、PNH、HS,血清中Hp的水平显著下降,Hp检测已作为临床溶血疾病诊断的常规指标。在感染、组织损伤过程中,Hp合成水平明显增强,反映了急性反应的状态[12]。Hp除参与炎性反应、溶血等,在肿瘤免疫抑制中也起重要的作用[13]。在肺癌、肝癌、卵巢癌等癌症患者的血清中具有较高的表达,此血清标志物联合其他肿瘤标志物对疾病早期特异性诊断有较大参考价值和意义[14]。随着研究的不断深入,发现Hp参与很多的生命活动,因此对Hp的生物信息学研究具有重要价值。
本研究通过生物信息学的手段对Hp基本生物信息进行预测分析,其结果表明,Hp不具有跨膜序列,是胞内蛋白,具有亲水性,无核定位序列,具有信号肽序列,主要在胞浆内行使功能。二级结构分析显示主要有β-折叠、α-螺旋、延伸链及无规则卷曲等组成。三级结构预测模型经验证也具有较高的稳定性,可信度较高。蛋白互作和GO注释结构显示Hp参与血管生成、炎性反应、抗菌作用及蛋白结合等过程,结合游离血红蛋白,并由CD163介导的内吞作用代谢。本研究通过生物信息学的方法得到的Hp的预测结构具有可靠性,对后期研究血清Hp参与的生理功能和机制研究具有指导意义,同时结合血红蛋白及结合其他肿瘤标志物对疾病的早期诊断和靶向药物的研究具有基础作用。
4 结语
随着生命科学及医学的发展,生物信息学作为一项重要的研究手段已经广泛的应用到结果分析及预测中,并在临床研究中发挥重要的作用。
