Targeted agents for second-line treatment of advanced hepatocellular carcinoma
2019-10-23NicolaPersoneniTizianaPressianiSilviaBozzarelliLorenzaRimassa
Nicola Personeni,Tiziana Pressiani,Silvia Bozzarelli,Lorenza Rimassa
Nicola Personeni,Tiziana Pressiani,Silvia Bozzarelli,Lorenza Rimassa,Medical Oncology and Hematology Unit,Humanitas Cancer Center,Humanitas Clinical and Research Center,IRCCS,Rozzano 20089,Milan,Italy
Nicola Personeni,Lorenza Rimassa,Department of Biomedical Sciences,Humanitas University,Pieve Emanuele 20090,Milan,Italy
Abstract
Key words: Hepatocellular carcinoma; Advanced-metastatic; Second-line; Third-line; Regorafenib; Cabozantinib; Ramucirumab; Angiogenesis; Multikinase inhibitor; MET;AXL; Vascular endothelial growth factor receptor 2
INTRODUCTION
Liver cancer ranks second among major causes of cancer-related deaths globally.In particular,hepatocellular carcinoma (HCC) roughly represents 90% of all primary liver cancers with 800000 new cases reported yearly[1].In more than 80% of patients,cirrhosis is a predisposing condition[2],often related to prior infection with hepatitis B virus (HBV),hepatitis C virus (HCV),or alcohol abuse.Less frequently,HCC may also arise in a non-cirrhotic liver as a consequence of HBV genotoxic effects,nonalcoholic steatohepatitis (in patients with metabolic syndrome and diabetes),or malignant transformation of a hepatocellular adenoma.
Even under rigorous surveillance programs,a sizeable proportion of patients are often diagnosed at a stage not amenable to potentially curative approaches[3],thereby prompting the search for palliative treatments.
In recent years,transcriptome analyses have allowed to increase our understanding of HCC complexity with the identification of a proliferation class and a nonproliferation one[2,4].Both classes display recurrent genetic alterations that affect deregulated pathways relevant to cellular homeostasis,senescence,proliferation,and differentiation.Although the ultimate goal of such advances is to inform future treatment strategies,none among driver mutations leading to oncogenic addiction in HCC is thus far considered as actionable[5].
On top of that,additional hurdles that hamper the development of personalized therapies lie within a substantial intra- and inter-tumor heterogeneity,that result from an admixture of mature hepatocytes and hepatic progenitor cells,both contributing to chronic inflammation,advanced fibrosis,and eventually cancer development[6].
Despite the obvious disappointment following the results of the first biomarkerdriven phase III trial ever done in HCC,that reported negative results for tivantinib in patients with MET-high HCC in 2018[7],the quest for personalized approaches is still underway within newer studies that may finally provide a conceptual frame for precision medicine in this hard-to-treat malignancy.Similar approaches could also take advantage from next generation sequencing (NGS) platforms that were recently presented as a useful tool to individualize available targeted therapies in HCC patients[8].Nevertheless,the clinical value of molecular profiling still needs to be demonstrated given that only few patients could receive targeted treatments matching with potentially actionable alterations identified by NGS[8].
Meanwhile,different strategies directed against relevant angiogenesis pathways have provided valuable therapeutic options in the management of advanced HCC.Indeed,a continuous dependence upon pro-angiogenic pathways is typical for HCC and it is reflected by an abnormal hypervascularity well known by the radiologist.This is mainly due to a hypoxic tumor microenvironment (TME) being a major determinant for hypoxia-inducible factor-1 transcription,that in turn leads to the over-production of vascular endothelial growth factor (VEGF).From a clinical standpoint,these peculiar aspects render HCC rather unique in comparisons with other cancers[9]and have long been proven useful for either HCC diagnosis or embolization therapies.
More than a decade ago,the approval of the multikinase inhibitor sorafenib has paved the way of anti-angiogenic therapies targeting VEGF and the VEGF receptors(VEGFRs) in the treatment of advanced HCC patients,not amenable to curative treatments[10].In addition to sorafenib,the therapeutic armamentarium for the frontline setting has been recently expanded with lenvatinib,which is a different multikinase inhibitor,still retaining anti-angiogenic properties.As reported in the overall survival analysis (OS) of the REFLECT trial,lenvatinib is non-inferior to sorafenib in untreated patients with advanced HCC[11].
However,intolerance or resistance to frontline sorafenib (or lenvatinib) may become major issues eventually leading to treatment failure.Whereas sorafenib targets encompass both drivers of cancer cell proliferation and the TME,such pharmacological complexity has greatly hampered the search for predictive markers and,arguably,the identification of resistance mechanisms.Nevertheless,after a decade with unsatisfactory results,three novel compounds sharing peculiar VEGFRs inhibition profiles have been recently reported superior when compared to placebo for OS in the second-line setting.These include regorafenib,cabozantinib (both belonging to the multikinase inhibitors class) and ramucirumab (a monoclonal antibody that targets VEGFR 2 signaling).
Aim of this review is to summarize current knowledge on the aforementioned agents and their role in the treatment of HCC patients who failed or are intolerant to sorafenib.
Regorafenib
Regorafenib is an orally administered tyrosine-kinase inhibitor that blocks the activity of several receptors such as VEGFR 1,2,and 3,tyrosine-protein kinase receptor,and platelet-derived growth factor receptor β[12].Based on the crucial role of angiogenesis in HCC development and progression,and on the results of a phase II study in patients with well-preserved liver function (Child-Pugh class A) progressing on sorafenib[13],regorafenib has been investigated in a large international phase III trial(ClinicalTrials.gov NCT01774344)[14].
The RESORCE trial was a multicenter,randomized,double-blind,placebocontrolled phase III trial assessing the role of regorafenib in patients affected by HCC progressing on sorafenib.Principal inclusion criteria were Barcelona clinic liver cancer(BCLC) stage B or C,preserved liver function (Child-Pugh class A),Eastern Cooperative Oncology Group (ECOG) performance status (PS) 0 or 1.Pathologic confirmation of diagnosis was not mandatory for patients with confirmed cirrhosis.Patients should have been treated with sorafenib at a minimum dose of 400 mg daily for at least 20 of the 28 d before discontinuation and should have stopped sorafenib no more than 10 wk before randomization.Reason for sorafenib discontinuation had to be documented radiologic progression,while patients intolerant to sorafenib or who had stopped sorafenib due to toxicity were not allowed to be included in the trial.
The trial randomized 573 patients from May 2013 to December 2015.Patients were randomized in a 2:1 ratio to receive regorafenib (n= 379) or placebo (n= 194) and were stratified by geographical origin (Asiavsrest of world),ECOG PS (0vs1),blood alpha-fetoprotein (AFP) levels (< 400 ng/mLvs≥ 400 ng/mL),extrahepatic disease(yesvsno),and macrovascular invasion (yesvsno).
Patients in the two groups were well-balanced for baseline characteristics,including sex,race,geographical origin,stage of disease,stage of liver dysfunction,and etiology.The proportion of patients with Asiatic origin was 38%.The treatment consisted in four 40 mg tablets of regorafenib (160 mg) orally or matching placebo once daily for 21 consecutive days,followed by 7 d of rest in 4-wk cycles.Treatment could be interrupted for disease progression according to modified RECIST(mRECIST)[15],clinical progression,death,unacceptable toxicity,or decision by the investigator.Tumor assessments were performed every 6 wk for the first 8 cycles and every 12 wk thereafter.The primary end-point of the study was OS in the intent-totreat population (ITT).Secondary endpoints were progression-free survival (PFS),time to treatment progression (TTP),objective response rate (ORR),and disease control rate (DCR) assessed by the investigators using mRECIST and RECIST v.1.1[16].Further endpoints were safety,pharmacokinetics (PK),biomarker evaluation,and quality of life (QOL).
At the data cut-off for final analysis (February 29,2016),among patients who started treatment (n= 567),309 (83%) in the regorafenib arm and 183 (95%) in the placebo arm discontinued the study drug.The most frequent reason for treatment discontinuation was disease progression.Median treatment duration was 3.6 mo with regorafenib and 1.9 mo with placebo.With a median follow-up of 7 mo,median OS was 10.6 mo in the regorafenib armvs7.8 mo in the placebo arm [hazard ratio (HR) =0.63 95% confidence interval (CI):0.50-0.79,P< 0.0001].The same survival gain was confirmed in the updated survival analysis performed almost 1 year after the first one(10.7 movs7.9 mo,HR = 0.61,P< 0.0001)[17].Median PFS by mRECIST was 3.1 mo in regorafenib arm and 1.5 mo in the placebo arm.Regorafenib was superior to placebo in all the efficacy endpoints and similar results have been demonstrated by RECIST 1.1 assessment (Table1)[18].
The safety population included 567 patients (99% of randomized patients),374 in the regorafenib group and 193 in the placebo group.All patients in the regorafenib arm and 93% of patients in the placebo arm had at least one adverse event (AE),graded using NCI-CTCAE version 4.03.These AEs were deemed related to the study drug in 93% of patients on regorafenib and 52% of patients on placebo (Table2).Most frequently observed grade 3-4 AEs were hypertension (15% of patients on regorafenibvs5% of patients on placebo),hand-foot skin reaction (HFSR) (13%vs1%),fatigue (9%vs5%),and diarrhea (3%vsnone).According to prior sorafenib dosing,grade ≥ 3 HFSR,fatigue,anorexia,and increased bilirubin were slightly higher in the group of patients that received < 800 mg compared with 800 mg,as last dose,while no difference was observed in rates of other treatment-emergent adverse events (TEAEs).Therefore,the last sorafenib dose may not predict the onset of TEAEs occurring with regorafenib[19].Serious AEs (SAEs) and death rates were similar in the two groups;SAEs were attributed to the study drug in 10% of patients on regorafenib and 3% of patients on placebo.Grade 5 AEs occurring within 30 d after the last dose of treatment were observed in 13% of patients in regorafenib patients and 20% in placebo arm and were deemed related to the study drug in 7 patients on regorafenib and 2 patients on placebo (both liver failure).Regorafenib was interrupted or reduced in 68% of patients and discontinued in 25% of patients due to AEs,while 31% of patients on placebo interrupted or reduced treatment and 19% of patients on placebo discontinued due to AEs.The most common AEs responsible for regorafenib discontinuation were aspartate aminotransferase (AST) or alanine aminotransferase increase (2% and 1%)and HFSR (2%).
Quality of life during the study was assessed by several questionnaires (Functional Assessment of Cancer Therapy-General and Hepatobiliary,EQ-5D,EQ-VAS) and no statistically significant changes in QOL were detected between the two treatment arms.
Further exploratory data showed that the sequence of sorafenib followed by regorafenib achieved a median OS of 26 mo[19].The efficacy of regorafenib was assessed according to the pattern of progression on prior sorafenib[20]and to the last sorafenib dose[19].Regorafenib was shown to provide significant survival benefits regardless of the pattern of progression and the last sorafenib dose,although the development of new distant metastases or vascular invasion was confirmed to be a negative prognostic factor.Furthermore,a negative correlation between baseline AFP and circulating MET levels and prognosis was confirmed regardless of treatment[21].Of note OS with regorafenib was significantly higher in patients suffering from HFSR[22],and this is in line with the correlation between skin toxicity and prognosis prospectively demonstrated with sorafenib[23].
In subsequent analyses regorafenib population PK (popPK) and exposure-response relationship were studied.PopPK analysis showed that most intrinsic factors had no statistically significant or clinically relevant impact on regorafenib exposure.Only age was found to be related to differences in exposure but the impact on efficacy was considered not significant[24].No statistically significant correlations between exposure and outcomes were identified[25].
Preplanned,retrospective,optional biomarker analyses on archival tumor tissues and plasma samples collected at baseline were performed to identify potential biomarkers correlating with clinical outcome[26].Baseline patient and disease characteristics were similar in the overall RESORCE population and in the plasma biomarker analysis cohorts,while several differences were reported between the overall study population and the tumor biomarker analysis cohorts due to the small sample size.Out of the 573 patients enrolled,only 68 archival tumor samples were collected while plasma samples were available for all the enrolled patients.For the NGS analysis,23 tumor samples (all in the regorafenib arm) were selected,and 17 were of sufficient quality for analysis.For the immune profiling analysis,62 samples had sufficient RNA,and 46 were of sufficient quality for analysis (32 in the regorafenib arm,14 in the placebo arm).The NGS analysis showed mutations inCTNNB1in 3/10 progressors and 0/7 responders,andVEGFAamplification in 1/7 responders and 0/10 progressors.The immune profiling analysis defined immune gene expression signatures with 3 groups with low (46%),medium (37%),and high(17%) immune cell scores.However,the small sample size precluded any meaningful conclusions and these results can be considered only hypothesis generating.For theplasma analyses,499 samples were of sufficient quality for protein analysis (332 in the regorafenib arm,167 in the placebo arm),and 343 were of sufficient quality for RNA analysis (234 in the regorafenib arm,109 in the placebo arm).The plasma analyses revealed multiple proteins and miRNAs possibly predictive for OS in patients treated with regorafenib.In particular,they identified 5 OS predictive biomarkers(angiopeietin-1,cystatin B,the latency-associated peptide of transforming growth factor beta1,oxidized low density lipoprotein receptor 1 C-C motif chemokine ligand 3),and 47 TTP predictive biomarkers[26].Finally,an exploratory analysis on 328 whole blood DNA samples identified single nucleotide polymorphisms (SNPs) prognostic for TTP,one of which,in theUGT1A1gene,was also predictive of regorafenib TTP benefit.Also,two SNPs in theVEGFAgene were identified as having a prognostic or predictive treatment effect on grade ≥1 HFSR[27].

Table1 Efficacy results of the RESORCE phase lll trial
Based on the results of the phase III RESORCE trial,regorafenib has been approved by the United States Food and Drug Administration (FDA),the European Medicines Agency (EMA),and many other regulatory agencies for the treatment of patients with advanced HCC previously treated with sorafenib.The recommended dose and schedule for regorafenib in HCC is 160 mg administered orally once daily for 21 d every 28 d.As mentioned above,prior sorafenib tolerance and preserved liver function (Child-Pugh class A) remain crucial to determine the eligibility status for treatment with regorafenib.
Cabozantinib
Cabozantinib is an oral multikinase inhibitor of several receptors including MET,VEGFR 1,2,3,AXL (GAS6 receptor),and RET.Other known targets of cabozantinib include ROS1,TRKA,TRKB,TYRO3,MER,KIT,and FLT-3[28].Based on preclinical studies in HCC models demonstrating the role of VEGFRs,MET,and AXL in tumorprogression[29],of MET in acquired resistance to antiangiogenic therapy including sorafenib[7,30-32],and on the results of a phase II randomized discontinuation trial in HCC[33],cabozantinib has been tested in the multicenter,randomized,double-blind,placebo-controlled phase III CELESTIAL trial (ClinicalTrials.gov NCT01908426)[34].The CELESTIAL trial enrolled patients with pathologic diagnosis of HCC not amenable to curative treatment,preserved liver function (Child-Pugh class A),and good PS (ECOG 0 or 1).Enrolled patients had received previous treatment with sorafenib,they could have received up to two prior systemic regimens for advanced HCC and had progressed following at least one prior systemic treatment.From September 2013 to September 2017,773 patients were randomized.At the time of the second interim analysis of OS (data cutoff of June 1,2017) 707 patients were randomized (2:1 ratio) to receive cabozantinib (n= 470) or placebo (n= 237) and constitute the ITT population for efficacy analyses.Randomization was stratified by disease etiology (HBV with or without HCVvsHCV without HBVvsother),region(Asiavsother),macrovascular invasion and/or extrahepatic disease (yesvsno).Baseline patient characteristics were well-balanced between the two treatment arms.All patients had received prior treatment with sorafenib,and 192 patients (27%) had received two previous systemic therapies for advanced HCC.Patients received 60-mg cabozantinib tablets or matching placebo once per day continuously.Tumor assessment was performed every 8 wk according to RECIST 1.1[16].Patients received treatment until loss of clinical benefit (treatment beyond radiographic progression was allowed) or unacceptable AEs.The primary endpoint was OS in the ITT population,secondary endpoints were PFS and ORR assessed by the investigators using RECIST 1.1[16].At the time of data cutoff,73 patients (16%) in the cabozantinib arm and 26 patients (11%) in the placebo arm were still on treatment.The most common reason for discontinuation was radiographic disease progression.One hundred and twenty-three patients (26%) in the cabozantinib arm and 78 patients(33%) in the placebo arm received post-study systemic or liver-directed therapy.Median OS was 10.2 mo (95%CI:9.1-12.0) in the cabozantinib armvs8.0 mo (95%CI:6.8-9.4) in the placebo arm,with a HR of 0.76 (95%CI:0.63-0.92) and aPvalue of 0.005.This value met the criterion for statistical significance at the second interim analysis(stopping boundaryP= 0.02),which included 484 deaths,corresponding to 78% of the 621 deaths planned for the prespecified final analysis.Cabozantinib performed better
than placebo in all the efficacy endpoints (Table3)[34-35].Preplanned and exploratory analyses confirmed the superiority of cabozantinib compared to placebo in all subgroups of patients.In the subgroup of patients who had received only sorafenib as previous systemic treatment,median OS was 11.3 mo in the cabozantinib group and 7.2 mo in the placebo group (HR = 0.70,95%CI:0.55-0.88),and median PFS was 5.5 mo in the cabozantinib group and 1.9 mo in the placebo group (HR = 0.40,95%CI:0.32-0.50).Cabozantinib improved clinical outcomes irrespective of prior sorafenib duration[36],age (cutoff 65 years)[37],baseline AFP values[38],prior transarterial chemoembolization (TACE)[39],tumor burden[40],and in patients with HBV etiology[41].Furthermore,47% of patients on cabozantinib compared to 11% of patients on placebo had any reduction in target lesions,and among patients with elevated baseline AFP levels,23% of patients on cabozantinib compared to 5% of patients on placebo achieved ≥ 50% reduction in AFP levels[35].AFP response rate,defined as ≥ 20%decrease in AFP level from baseline at week 8,was higher with cabozantinibvsplacebo and was associated with longer OS and PFS with cabozantinib[42].Although different cutoffs were adopted,these findings are in line with previous reports suggesting a benefit from systemic treatments in patients achieving an AFP response[43].

Table2 Adverse events in the RESORCE phase lll trial occurring in ≥ 10% of patients-Safety population
The safety population included 704 patients who started treatment,467 in the cabozantinib group and 237 in the placebo group.Median duration of treatment was 3.8 mo with cabozantinib and 2 mo with placebo.Ninety-nine percent of patients who received cabozantinib and 92% of patients who received placebo had ≥ 1 AE (graded according to NCI-CTCAE version 4.0),and 68% of patients on cabozantinib and 32%of patients on placebo had ≥ 1 grade 3-4 AE.Most common grade 3-4 AEs were palmar-plantar erythrodysesthesia (PPE) (17% of patients on cabozantinibvs0% of patients on placebo),hypertension (16%vs2%),increased AST level (12%vs7%),fatigue (10%vs4%),and diarrhea (10%vs2%) (Table4).SAEs were reported in 50% of patients in the cabozantinib arm and in 37% of patients in the placebo arm.Grade 5 AEs occurring within 30 d after the last dose of treatment,mostly disease progression,were observed in 12% of patients in both arms and were deemed related to the study drug in 6 patients on cabozantinib and in 1 patient on placebo.Dose reductions (to 40 mg and then to 20 mg daily) and discontinuations due to AEs occurred in 62% and 16% of patients on cabozantinib and in 13% and 3% of patients on placebo,respectively.AEs leading to treatment discontinuation in > 1.0% of patients in the cabozantinib group were PPE,fatigue,decreased appetite,diarrhea,and nausea.Median average daily dose was 35.8 mg for cabozantinib and 58.9 mg for placebo,and median time to first dose reduction was 38 d in the cabozantinib arm[34].The safety results for cabozantinib reported in the exploratory analyses were consistent with the results in the overall study population[36-41].Of note,albeit patients ≥ 65 years more frequently discontinued treatment due to AEs,rate of dose reductions and median average daily dose were similar irrespective of age[37].Grade 3-4 AEs were similar for HBV-positive patients and for patients with prior TACE compared to the overall study population and to patients without prior TACE,respectively[39-41].Also,a post hoc QOL analysis estimated the incremental quality-adjusted life-years (QALYs)accrued with cabozantinib.Although cabozantinib was associated with an initial,small reduction in health utility compared to placebo,the difference reduced with dose adjustments and considering the overall within-trial health utility experience,cabozantinib was associated with a clinically and statistically significant benefit in mean QALYs[44].Finally,a popPK analysis showed that PK of cabozantinib in HCC patients was similar to that observed for other cancer types and healthy volunteers,and that HCC patients with mild and moderate hepatic dysfunction had consistent exposure with the patients of normal liver function[45].
Based on the results of the phase III CELESTIAL trial,cabozantinib has been approved by the EMA and the FDA for the treatment of patients with HCC who have been previously treated with sorafenib.The recommended dose and schedule for cabozantinib in HCC is 60 mg,administered orally once daily (tablet formulation).
Given a strong preclinical rationale showing the effect of cabozantinib on immunemediated killing of tumor cells and immune TME permissiveness[46],ongoing studies are testing cabozantinib in combination with immune checkpoints inhibitors.Notably,the multicenter,randomized,open-label,controlled phase III COSMIC-312 trial(ClinicalTrials.gov NCT03755791) is evaluating the efficacy and safety of cabozantinib in combination with atezolizumabvsthe standard of care sorafenib in patients with advanced HCC who have not received previous systemic therapy.
Ramucirumab
Ramucirumab is a recombinant human IgG1 monoclonal antibody that interferes with high affinity with the extracellular domain of VEGFR 2,blocking the binding of its ligands VEGF-A,VEGF-C,and VEGF-D,that play an important role in tumorangiogenesis and tumor growth[47].Two phase I trials evaluated ramucirumab in order to define the maximum-tolerated dose with doses ranging from 2 mg/kg per week to 20 mg/kg per 3 wk intravenously and two patients with advanced HCC experienced disease control longer than 6 mo[47-48].These results provided the rationale for a phase II study that confirmed the antitumor activity with an acceptable safety profile of ramucirumab 8 mg/kg per 2 wk in first-line HCC[49].The phase III REACH trial(ClinicalTrials.gov NCT01140347) evaluated ramucirumab 8 mg/kg per 2 wkvsplacebo in 565 patients with advanced HCC as second-line treatment following sorafenib.With a median OS of 9.2 mo in the ramucirumab arm and of 7.6 mo in the placebo arm (HR = 0.87,95%CI:0.72-1.05,P= 0.14),this trial did not meet its primary endpoint.However,in the prespecified analysis of the subgroup of patients with baseline AFP levels ≥ 400 ng/mL (n= 250),ramucirumab showed a significant survival benefit,with a median OS of 7.8 movs4.2 mo (HR=0.67,P=0.006),with a good safety profile.In addition,the REACH trial confirmed in the overall study population the negative prognostic role of baseline elevated AFP levels[50].
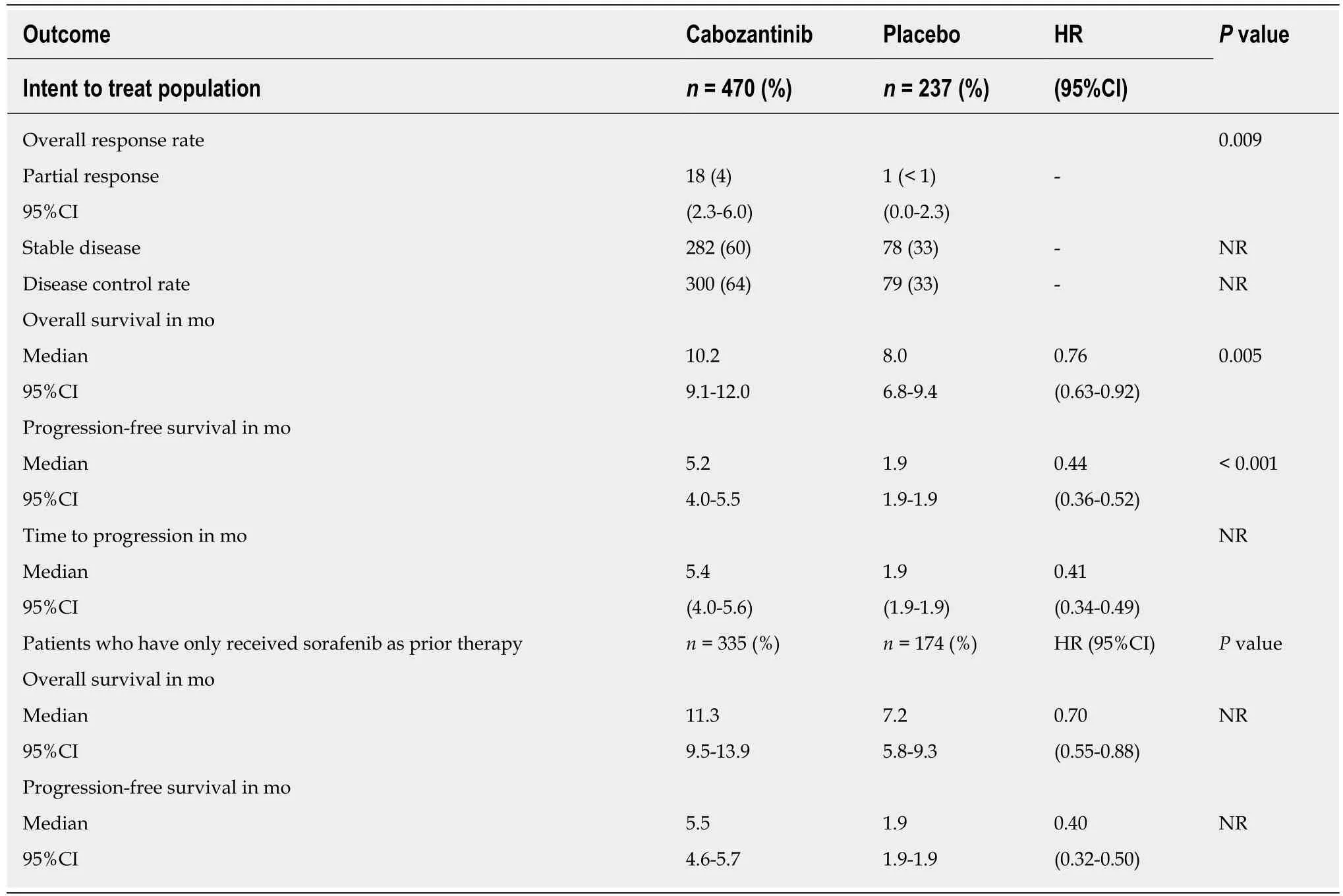
Table3 Efficacy results of the CELESTlAL phase lll trial
In HCC the AFP value is included in several prognostic scoring systems[51-53]and a concentration > 400 ng/mL has been associated with worse prognosis[50,54].Also,increased VEGFR expression and angiogenesis have been demonstrated in patients with HCC and elevated AFP concentration[2,55,56].
Based on these data and on the results achieved in the REACH trial in patients with high baseline AFP levels,ramucirumab was further tested in the phase III multicenter,randomized,double-blind,placebo-controlled REACH-2 trial (ClinicalTrials.gov NCT02435433)[57].
The REACH-2 trial enrolled patients with histologic or cytologic diagnosis of HCC or,in the absence of histologic confirmation,with cirrhosis and HCC,BCLC stage B,or C disease not suitable for locoregional therapy,preserved liver function (Child-Pugh class A),and good PS (ECOG 0-1),AFP levels ≥ 400 ng/mL,progressing on or intolerant to first-line treatment with sorafenib.From July 26,2015,to August 30,2017,292 patients were randomly assigned (2:1 ratio),197 to the ramucirumab group and 95 to the placebo group.Randomization was stratified by geographical region[region 1 (Americas,Europe,Australia,Israel)vsregion 2 (Asia,excluding Japan)vsregion 3 (Japan)],macrovascular invasion (yesvsno),and ECOG PS (0vs1).Patientsreceived ramucirumab 8 mg/kg or placebo intravenously every 14 d until disease progression,unacceptable toxicity,or withdrawal of consent.Tumor assessment was performed by the investigators every 6 wk according to RECIST 1.1 during the first 6 mo of treatment,and every 9 wk thereafter.Patient-reported outcomes were assessed at baseline,every 6 wk,and at treatment discontinuation with the Functional Assessment of Cancer Therapy Hepatobiliary Symptom Index 8 (FHSI-8),which specifically evaluates the most frequently observed symptoms of patients with hepatobiliary malignancies:lack of energy,nausea,pain,weight loss,back pain,fatigue,jaundice,and stomach pain or discomfort[58].Serum AFP levels were measured at baseline,every 6 wk during the treatment period,and at the end of the treatment period.
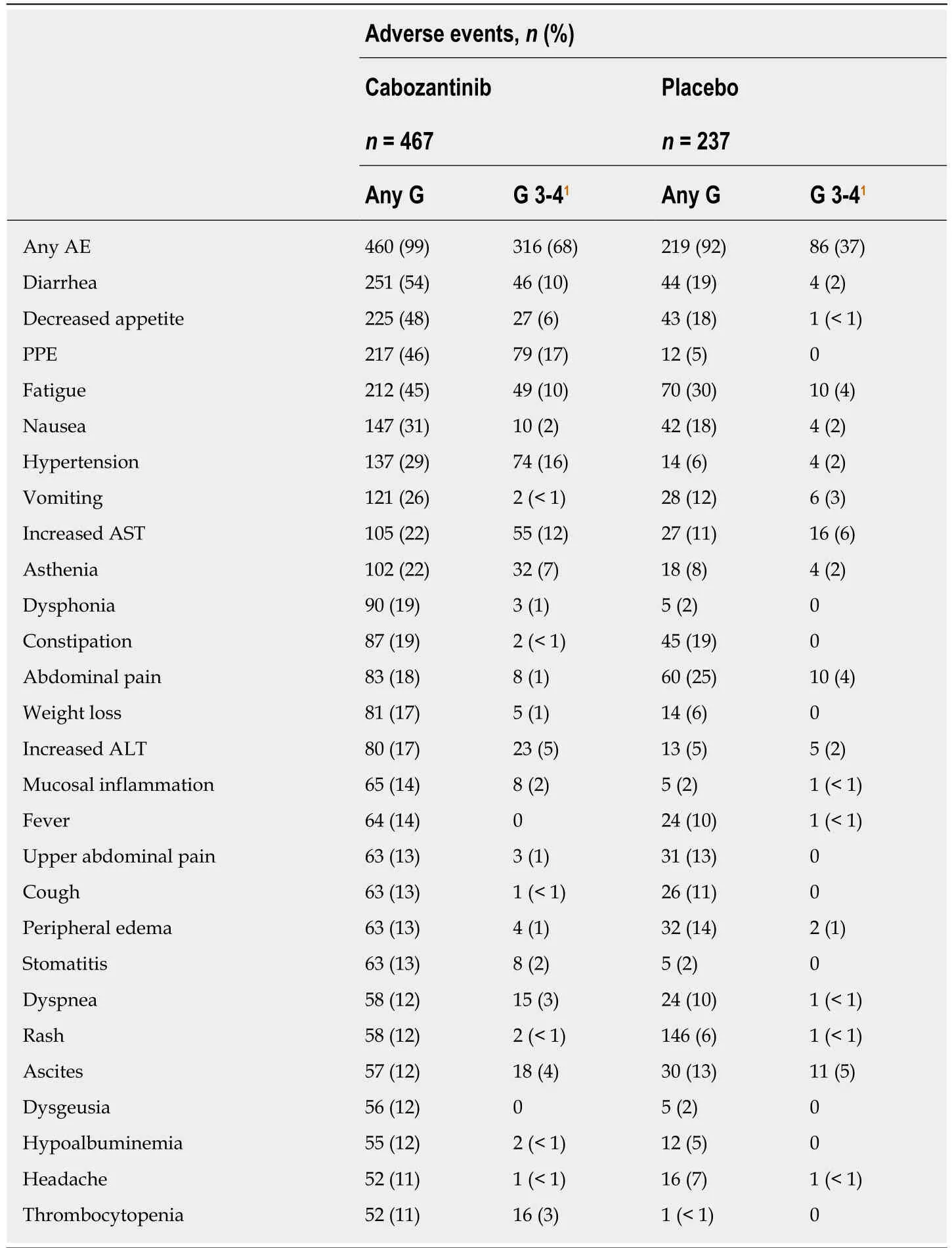
Table4 Adverse events in the CELESTlAL phase lll trial occurring in ≥ 10% of patients regardless of causality-Safety population
The primary endpoint of REACH-2 trial was OS,secondary endpoints were PFS,ORR,TTP,safety,time to deterioration in scores on the FHSI-8,and time to deterioration in ECOG PS.Efficacy analyses were conducted by ITT,safety analyses were done in all patients who received at least one dose of study drug.Baseline patient characteristics were well-balanced between the two treatment arms,except for baseline AFP levels that were higher in the ramucirumab group [2741 ng/mL (IQR 1178-11861) in the placebo groupvs3920 ng/mL (IQR 1175-20000) in the ramucirumab group].Median duration of prior sorafenib was 4.1 mo in both groups,and 50 patients (17%) discontinued sorafenib due to intolerance.At the time of data cutoff,March 15,2018,281 patients were off treatment,and 11 patients in the ramucirumab group were still receiving therapy; 206 patients (71%) had disease progression,and 221 (76%) had died.Median OS was 8.5 mo (95%CI:7.0-10.6) in the ramucirumab armvs7.3 mo (95%CI:5.4-9.1) in the placebo arm,with a HR of 0.71(95%CI:0.53-0.95) and aPvalue of 0.0199.Ramucirumab also significantly improved PFS (2.8vs1.6 mo,HR = 0.45,95%CI:0.339-0.603,P< 0.0001),and DCR (59.9%vs38.9%,P= 0.0006).These results were confirmed in almost all predefined subgroups(Table5).
Median duration of treatment was 12 wk with ramucirumab and 8 wk with placebo.Treatment discontinuations due to any AEs (graded according to NCICTCAE version 4.0) (18%vs11%) and to treatment-related AEs (11%vs3%) of any grade were more frequent in the ramucirumab group compared to the placebo group.Dose reductions (5%vs2%),delays (6%vs3%),and omissions (29%vs11%) due to AEs were more common in the ramucirumab arm than in the placebo arm.Most common AEs of any grade in the ramucirumab group were fatigue (27%),peripheral edema (25%),hypertension (25%),and decreased appetite (23%).Hypertension (12%vs4%) and hyponatremia (5%vs2%) were the only grade ≥ 3 AEs reported in ≥ 5% of patients (Table6).SAEs occurred in 35% of patients in the ramucirumab group and 29% in the placebo group.Grade 5 AEs occurring within 30 d after the last dose of treatment were observed in 20% of patients in the ramucirumab arm and 17% of patients in the placebo arm,and were deemed related to the study drug in 4 patients on ramucirumab (3 liver failure and 1 arterial thromboembolic event) and in no patient on placebo.FHSI-8 was completed in 99% of patients at baseline and 67% at the end of treatment in both groups.Median time to deterioration of FHSI-8 scores(3.7 movs2.8 mo,P= 0.238) and ECOG PS (P= 0.77) were not different between the ramucirumab and placebo arms,although the number of events did not allow a meaningful statistical assessment of ECOG PS deterioration[58].
An exploratory analysis investigated the potential relationship between changes in AFP during treatment and efficacy in terms of survival,considering AFP response defined as ≥ 20% decrease from baseline.Ramucirumab was shown to prolong time to AFP progression and radiographic TTP and to slow the rate of AFP increase compared to placebo.AFP response was significantly higher in the ramucirumab arm compared to the placebo arm (42%vs11%,P< 0.0001).Also,regardless of treatment OS was longer in patients with AFP response (13.5 mo in AFP respondersvs6.7 mo in non-responders,HR = 0.47,P< 0.0001),and changes in AFP levels were associated with radiographic TTP (3 mo in AFP respondersvs1.6 mo in non-responders,HR =0.43,P< 0.0001)[59].
A preplanned pooled meta-analysis of individual data of patients (n= 542) enrolled in the REACH-2 trial and patients with AFP ≥ 400 ng/mL enrolled in the REACH trial(ramucirumab,n= 316; placebo,n= 226) confirmed significant improvements in OS(median 8.1vs5.0mo with placebo,HR = 0.69,95%CI:0.57-0.84,P= 0.0002),PFS(median 2.8vs1.5mo with placebo,HR = 0.57,95%CI:0.47-0.69,P< 0.0001),and DCR(56.3%vs37.2% with placebo,P< 0.0001).Of note,median baseline AFP levels were lower in the REACH-2 trial compared to those in the above mentioned cohort of the REACH trial [3394 ng/mL (IQR 1177-16812)vs5736 ng/mL (IQR 1322-291000)][58].The same pooled analysis showed a reduction in disease-related symptoms with ramucirumab compared to placebo,with a significantly delay in FHSI-8 time to deterioration (3 mo with ramucirumabvs1.9 mo with placebo)[60].Also,the pooled analysis confirmed the efficacy and safety results regardless of etiology,including HCV,HBV,and other[61],and in Japanese patients[62].Finally,an exploratory analysis evaluated the prognostic utility of Child-Pugh scorevsalbumin-bilirubin (ALBI)grade,showing a similar prognostic utility of the two scoring system and a higher incidence of liver AEs in patients with a high score in either system,and confirming the efficacy of ramucirumab in patients with ALBI score 1 or 2 or Child-Pugh score 5 or 6[63].
Based on these results ramucirumab,pending approval,will be a new treatment option for patients previously treated with sorafenib and with baseline elevated AFP levels.The recommended dose and schedule for ramucirumab is 8 mg/kg intravenously every 14 d.
CONCLUSION
Despite numerous negative trial results in the second-line setting,the current clinical scenario is quickly expanding with the anticipated availability of three antiangiogenic agents-regorafenib,cabozantinib,and ramucirumab-shown to prolong OS in recent phase III second-line trials.In contrast,preliminary data from a phase III trial of pembrolizumab[64]suggest that single-agent immune checkpoint inhibitors mightnot be superior to placebo in patients who had received prior sorafenib.In fact,despite numerically longer OS and PFS in the pembrolizumab arm of the KEYNOTE-240 study,the statistical significance per pre-specified statistical plan was not met.Pending additional details from KEYNOTE-240 that need to be considered,these disappointing results do not necessarily imply a dead end for immunotherapy studies in HCC.

Table5 Efficacy results of the REACH-2 phase lll trial
Rather,combinations of immune checkpoint inhibitors and anti-angiogenics may represent a sound evolution of current treatment options.In this respect,it is predicted that some agents may also move from the second-line to a frontline setting,as it is the case for current phase I trials of regorafenib plus pembrolizumab(ClinicalTrials.gov NCT03347292),or cabozantinib plus nivolumab as neoadjuvant treatment (ClinicalTrials.gov NCT03299946).In principle,robust preclinical data do support similar strategies aiming to improve the effectiveness of immunotherapy converting an immunosuppressive milieu into an immunosupportive one[65].However,most clinical studies are still in their very early stages of development,while other studies are already making their way into more advanced phase III contexts (ClinicalTrials.gov NCT03755791).
For the time being,in the absence of additional agents looming in the spotlight of placebo-controlled studies,an anti-VEGFR strategy is overall regarded as the only one increasing survival,and thereby establishing a standard of care after prior sorafenib treatment.Still,when it comes to specific treatment choices,the debate remains open as no direct comparative studies testing regorafenib,cabozantinib,and ramucirumab are available.With the notable exception of ramucirumab (whose efficacy could not be proven in patients with low AFP levels),there is no approved biomarker that can aid patient selection,and this observation clearly speaks to the huge translational efforts needed.As in other oncology settings,clinical factors informing treatment selection should include first-line therapy,tolerance,and duration of response to prior treatment.In fact,inclusion and exclusion criteria provided by each clinical study protocol should be an additional aid for the selection of the most adequate second-line agent.For instance,poor tolerability of prior sorafenib excludes an individual patient from treatment with regorafenib.Similarly,low AFP levels clearly contraindicate ramucirumab.In view of a treatment sequencing that includes up to three lines,consistent with CELESTIAL study[34],one may consider cabozantinib as a third-line treatment.Even patients’ clinical conditions by the time of disease progression,liver function,and the adverse events profiles are variables that need to be considered in the decision-making process.
Further,will the results of well-conducted clinical trials fulfill the expectations of the real-world setting? This is not a trivial point,and this will undoubtedly pose additional questions,especially in light of a limited benefit of sorafenib previously reported in a cohort of Medicare beneficiaries[66].
The scientific community is witnessing a turning point for our knowledge of thegenetic and immunologic landscape of HCC.Encouraging efficacy signals are now emerging from the use of second-line anti-angiogenic agents after sorafenib.Arguably,from a clinical perspective,the next challenge will be the implementation of well-designed studies that include sound correlative translational investigations.This is a great opportunity to bridge the enormous gap between clinical practice and basic science still existing in the field of HCC research.

Table6 Adverse events in the REACH-2 phase lll trial occurring in ≥ 10% of patients-Safety population
杂志排行

World Journal of Gastrointestinal Oncology的其它文章
- Cancer-specific metabolism:Promising approaches for colorectal cancer treatment
- Prognostic and pathological impact of tumor budding in gastric cancer:A systematic review and meta-analysis
- Race,the microbiome and colorectal cancer
- Precision medicine in gastric cancer
- Endoscopic management of esophageal cancer
- Retrospective review of total neoadjuvant therapy