[DMAPH]Br催化CO2和环氧氯丙烷反应的机理
2019-10-14宋振艳冯华杰李志伟
宋振艳,冯华杰,李志伟
(1.肇庆学院 环境与化学工程学院,广东 肇庆 526061;2.海南师范大学 化学与化工学院,海南 海口 571158)
近年来CO2加剧排放引发的全球变暖和环境恶化问题受到越来越多的关注,将大气中CO2固定转化为高附加值精细化学品,无论从资源回收利用还是从环境保护方面,都具有巨大的现实意义和应用前景.与其他常见碳源相比,CO2无毒价廉、取之不尽,被广泛应用于基础研究和应用研究领域[1-2].但相对于其巨大的储量,CO2的利用非常有限,原因在于它很高的热力学稳定性和动力学惰性,需要高效的催化剂才能将其活化.
在CO2固定活化催化剂中,离子液体(IL)有着举足轻重的地位[3-6].离子液体催化具有活性高、工艺简单、收率高、能循环利用、环境友好的优点.环碳酸酯是CO2固定转化中最流行的目标产物之一[7-9],为更深入地理解此类反应,以便开发高效、价廉、绿色、环保的离子液体催化体系,有必要对催化反应机理、反应途径,离子液体在反应中的作用进行剖析[10-11].最近,Zhang[12]等报道了离子液体[DMAPH]Br催化CO2与环氧氯丙烷生成环碳酸酯的反应,产物收率和选择性都很高,为明确其反应机理,通过理论计算从微观角度进行探讨.
1 计算方法
首先在B3LYP/6-31+G(d,p)计算水平下,对反应过程中不同步骤的反应物、过渡态、中间体和产物进行结构优化,然后对结构进行频率计算,确保反应物、中间体、产物是稳定的,而过渡态有且只有一个合理的虚频.为确保所有过渡态的准确性,即可通过势能面连接相应的反应物和产物,用内禀反应坐标(IRC)方法对反应路径进行验证.此外,根据前线分子轨道理论分析反应物间的成键情况.所有的计算都在Gaussian 09[13]程序包中完成,能量均采用校正后的吉布斯自由能.
2 结果与讨论
2.1 反应机理
计算体系如图1所示,离子液体催化剂为4-二甲胺基吡啶氢溴酸盐.首先提出2个反应途径A和B,反应物、中间体、过渡态及产物的结构分别如图2和图3所示,而能量如表1和图4所示,相对能量的零点为相同计算水平下所得的环氧氯丙烷、CO2以及催化剂[DMAPH]Br的能量之和.

图1 [DMAPH]Br催化CO2和环氧化合物的环加成反应
路径A中,环氧化物通过其氧原子与[DMAPH]Br的质子之间形成氢键而活化,导致C-O键极化,[DMAPH]Br中的Br攻击环氧化物中与氯甲基相连的碳原子(C1),经过相对能量为115.12 kJ/mol的过渡态TS1A,使其开环并形成中间体A1,这一步的活化能为70.64 kJ/mol,放热91.47 kJ/mol.在TS1A中,将要断键的O1-C1被拉长至0.171 4 nm,将要成键的O1-H1,Br-C1分别缩短至0.139 4和0.276 2 nm,TS1A有唯一虚频(-439.36 icm-1),其振动方式为O1和C1伸长、H1和O1缩短、C1和Br缩短,正是断键和成键的方向,可见这一步骤的过渡态是正确的.
随后A1的O原子与CO2的C原子结合,越过75.52 kJ/mol的能垒,经过过渡态TS2A,形成中间体A2,这一步骤吸热67.39 kJ/mol.在TS2A中,O1与C3的距离缩短至0.178 2 nm,逐渐靠近成键,H1与O1的相互作用减弱,键长拉长至0.133 4 nm,该过渡态有且只有一个虚频(-317.07 icm-1),振动方式为O1和C1靠近成键、H1和O1远离断键的方向,最终生成中间体A2,成功捕获CO2分子,同时Br-C1键长增加,作用减弱.
中间体A2继续反应,经过过渡态TS3A,能垒为101.68 kJ/mol,闭环形成环状碳酸酯,放热45.92 kJ/mol.在TS3A中,Br-C1键长增加至0.257 4 nm,O2和C1的距离缩短至0.193 5 nm,且有唯一虚频(-418.47 icm-1),振动方式为Br与C1伸长断键、C1和O2靠近成键的方向,最终闭环生成产物,同时离子液体催化剂[DMAPH]Br复原.

表1 相关中间体的相对吉布斯自由能(ΔG)
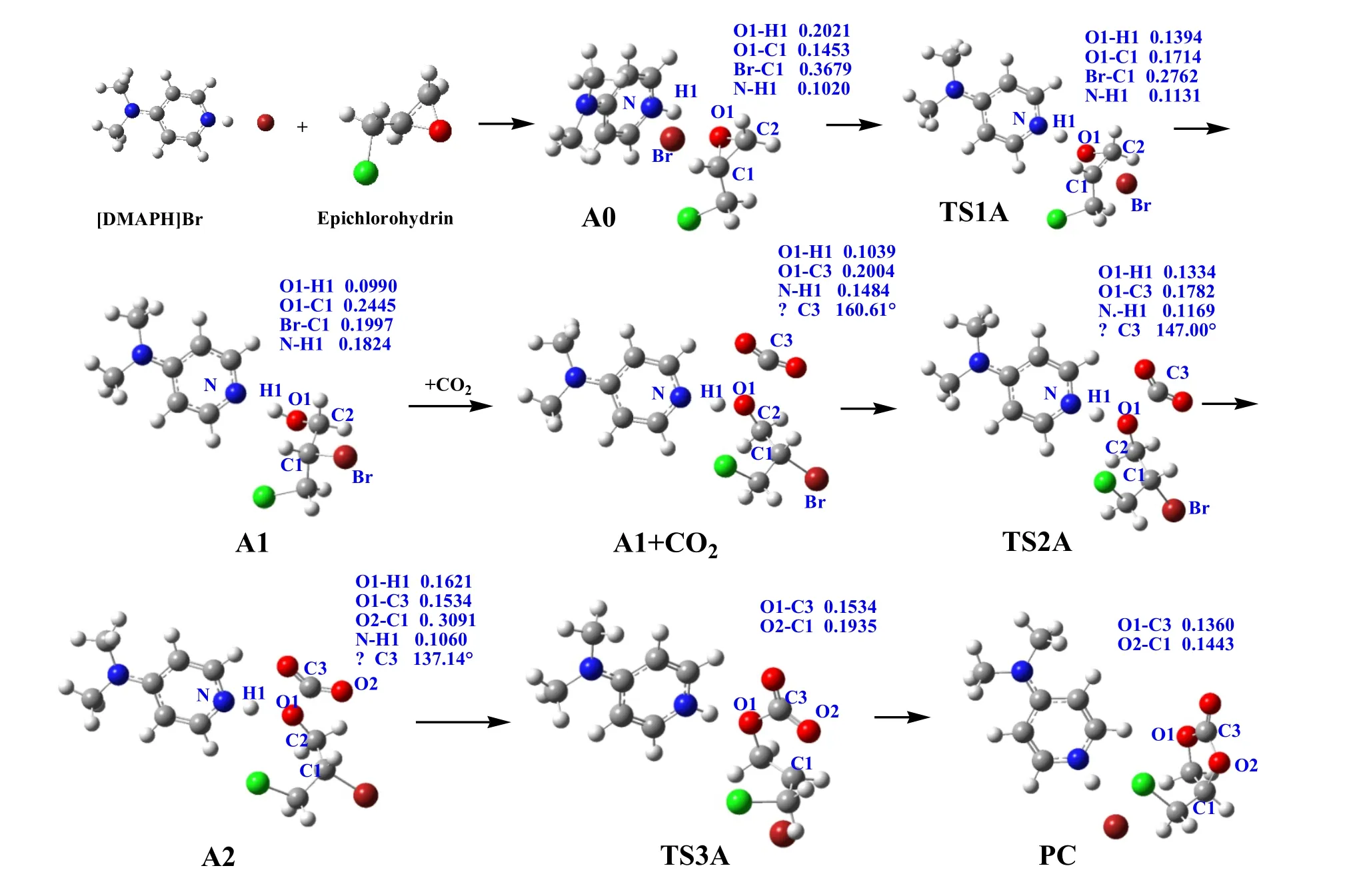
图2 反应路径A的反应物、中间体、过渡态和产物的结构和参数 (nm或 °)

图3 反应路径B的反应物、中间体、过渡态和产物的结构和参数 (nm或 °)
2.2 路径B的反应机理
与A不同,反应路径B中的Br攻击了环氧化物中与氧原子相连的另一个碳原子(C2).首先,由[DMAPH]Br的H原子使环氧氯丙烷中的氧原子活化,而Br离子亲核攻击C2原子,在2种因素的协同作用之下,经过能垒为74.44 kJ/mol的过渡态TS1B生成B1,放热95.46 kJ/mol.在TS1B中,H1和O1相互靠近,距离缩短至0.142 8 nm,Br与C2的距离缩短至0.280 5 nm,均有成键的趋势,而O1-C2键被拉长至0.165 5 nm,有断键趋势.过渡态TS1B有唯一虚频(-378.98 icm-1),且振动沿着H1与O1、Br与C2靠近成键,O1-C2键伸长断开的方向进行,最终生成B1结构,完成环氧氯丙烷的开环.

图4 路径A和B的相对吉布斯自由能图
然后,B1中插入CO2分子,经过能垒为79.07 kJ/mol的过渡态TS2B得到B2.TS2B中,被H1活化的O1逐渐远离H1,O1-H1伸长至0.137 1 nm,呈断键趋势.同时,O1与CO2分子的C3逐渐靠近,距离缩短至0.180 5 nm,CO2的空间结构也发生一定的变化,变为夹角为147.46°的非直线型,TS2B有且只有一个虚频(-246.11 icm-1),振动沿着O1-H1键拉长断键、O1和C3距离缩短成键的方向进行,分别指向TS2B前后的反应物和产物.经过过渡态TS2B结构生成B2,其相对能量为38.82 kJ/mol,完成CO2的捕捉,同时Br-C2键长增加,作用减弱.需要指出的是,如果不考虑零点能,则过渡态TS2B比中间体B2的能量高,考虑零点能后,B2的能量比TS2B略高,这是个较常见的问题[14-15].
第三步,中间体B2中的C2与CO2分子中的O2相互作用,经过过渡态TS3B,越过71.07 kJ/mol的能垒,闭环形成环状碳酸酯,放热53.29 kJ/mol.TS3B中,Br-C2键伸长至0.252 2 nm,呈断键趋势,而C2与O2距离靠近缩短至0.189 6 nm,呈成键趋势.过渡态TS3B有唯一虚频(-434.83icm-1),振动沿着旧键Br-C2断裂、新键C2-O2生成的方式进行,最终闭环完成反应,同时,离子液体催化剂[DMAPH]Br复原.
由上述分析和图4可知,路径A中,所有过渡态中TS3A相对能量最高,为133.13 kJ/mol,即路径A的开环、CO2插入、闭环3个步骤中,最后一步闭环生成产物所要跨越的能垒最高,是反应的决速步,而总的能垒为180.12 kJ/mol.路径B中,过渡态TS1B相对能量最高,为115.54 kJ/mol,即路径B开环、CO2插入、闭环3个反应步骤中,第一步开环生成B1所要跨越的能垒最高,是路径B的决速步,反应的总能垒为169.90 kJ/mol.由总能垒的大小可知,路径A的总反应能垒要比路径B总反应能垒高,因此反应路径B为更优反应路径.
2.3 前线分子轨道理论分析
图5给出了环氧氯丙烷、CO2、环氧氯丙烷和催化剂配合物A0和B0的前线轨道HOMO和LUMO以及相应的能量.从图5中可看出,电子从环氧氯丙烷的HOMO轨道跃迁到CO2的LUMO轨道所要跨越的能垒为818.29 kJ/mol,而电子从CO2的HOMO轨道跃迁到LUMO轨道所要跨越的能垒相对较高,为980.68 kJ/mol,但两者都很大,所以很难自发反应,需要催化体系来活化.

图5 前线分子轨道的能量(kJ/mol)
在催化剂的作用下,催化剂和环氧氯丙烷先形成配合物A0和B0,两者的LUMO和HOMO的能量相差不大,但与CO2相比则相差很大.根据前线分子轨道理论,A0和B0的LUMO与CO2的HOMO能量相差较大,分别为843.62和842.36 kJ/mol,而A0和B0的HOMO与CO2的LUMO能量相差较小,分别为527.95和527.05 kJ/mol,可见反应由A0和B0提供电子,CO2提供空轨道,且B0的HOMO与CO2的LUMO能量差与A0相比略低,从另一角度证实了B路径更优.
3 结论
在密度泛函理论水平B3LYP/6-31+G(d,p)下,计算了离子液体[DMAPH]Br催化CO2和环氧氯丙烷反应生成环碳酸酯的反应机理.设计了2个反应路径A和B,对不同路径所涉及的反应物、中间体、过渡态以及产物的相关结构参数和能量进行了理论计算.根据计算的结果,B路径的总能垒较低,所以路径B为更优反应路径.前线轨道理论分析表明:反应发生时环氧氯丙烷的HOMO轨道与CO2的LUMO轨道发生对称性重叠,电子从环氧氯丙烷的HOMO轨道流向CO2的LUMO轨道从而发生化学键的生成和断裂.
