基于标准制剂控制模式和定量指纹图谱评价复方甘草片的质量一致性
2019-10-11孙国祥迟晗笑孙万阳侯志飞李显林蒲道俊陈振鸿
闫 慧, 孙国祥*, 迟晗笑, 张 晶, 孙万阳, 侯志飞, 李显林, 蒲道俊, 陈振鸿
(1. 沈阳药科大学药学院, 辽宁 沈阳 110016; 2. 暨南大学中药和天然药物研究所, 广东 广州 510632; 3. 河北化工医药职业技术学院, 河北 石家庄 050026; 4. 国药集团工业有限公司, 北京 101300; 5. 西南药业股份有限公司, 重庆 400038; 6. 新疆富沃药业有限公司, 新疆 沙雅 842200)
复方甘草片是60多年前从美国引进的,原方出自苏格兰约翰·布朗医生(1735~1788)发明的布朗合剂(Brown Mixtura)[1],我国青海制药厂剔除方中酒石酸锑钾制成复方甘草片。每片复方甘草片中含甘草浸膏粉112.5 mg,阿片粉或罂粟果提取物粉4 mg,樟脑2 mg,八角茴香油2 mg,苯甲酸钠2 mg[2]。其中甘草浸膏粉为第一类主要药效物质,是保护性镇咳祛痰剂;阿片粉中的吗啡(morphine, MP)和磷酸可待因能有效镇咳,为第二类重要药效物质;樟脑及八角茴香油能刺激支气管黏膜,反射性地增加腺体分泌,稀释痰液,使痰易于咳出,此二原料为辅助药效物质;苯甲酸钠为防腐剂。因此,复方甘草片不但有镇咳祛痰作用,还对呼吸道黏膜具有一定的保护性作用[3,4]。复方甘草片同时具有镇痛作用,且大部分物质易透过血脑屏障,对阿尔兹海默症(AD)患者产生有益作用。《中国药典》要求测定吗啡和甘草酸(glycyrrhizic acid, CHA)含量以进行质量控制,文献通过测定复方甘草片中几种成分的含量来控制质量[5-8],但复方甘草片只通过几种物质含量来控制质量显然是不全面的。中药指纹图谱技术是一种整体定性和整体定量化技术手段,它能够从宏观上整体反映中药和植物药所含化学成分的种类和数量,是目前国际公认的中药或植物药的质量控制最佳手段[9-11]。因此本实验采用HPLC定量指纹图谱技术结合双标校正法,同时定量5个质量标志物,全面评价复方甘草片的质量。中药的质量标志物(quality marker, Q-marker)是指存在于中药材和中药产品(如中药饮片、中药煎剂、中药提取物、中成药制剂)中固有的或加工制备过程中形成的、与中药的功能属性密切相关的化学物质,作为反映中药安全性和有效性的标示性物质进行质量控制[12]。本文基于定量指纹图谱大数据筛选建立复方甘草片标准制剂控制模式,实现对其质量一致性评价和工艺一致性评价。
1 中药(植物药)质量一致性评价核心方法
1.1 中药(植物药)标准制剂控制模式
中药(植物药)标准制剂也称为中药(植物药)本底制剂,它是在中药(植物药)研制和创新过程中经过药效学和毒理学试验证明为最佳中药(植物药)组方(药效最佳和毒性最低)和具有恒定化学成分含量和分布比例的规范制剂,其主要指标成分含量变动范围在±5%内。中药(植物药)指纹图谱的宏定性相似度Sm不低于0.95,宏定量相似度Pm应在95%~105%,为标准制剂基础条件。标准制剂应诞生在新药创制期间,其可作为质量和药效对照物。现阶段已上市的每种中药(植物药)都固有一个标准制剂模式,需要分层取样并通过定量指纹图谱大数据考察获得标准制剂。基于标准制剂结合定量指纹图谱和多Q-markers的精准定量控制,构成中药(植物药)标准制剂控制模式(样品用标准制剂评价时:Sm不低于0.90,Pm应在80%~120%,同时指标成分含量合格,是合格品基本标志),具备直接用于中药(植物药)生产过程和终产品质量控制的基本功能。中药(植物药)不存在参比制剂,中药参比制剂必须经过从原料、中间体和制剂的标准化自证后即成为标准制剂。中药标准制剂控制模式为中药一致性评价提供了很好的思路。
1.2 系统指纹定量法(SQFM)

1.3 定量指纹图谱双标校正法

(1)
(2)
(3)
(4)
2 实验部分
2.1 仪器与试药
Agilent 1100型液相色谱仪(配有二极管阵列检测器、四元低压梯度泵、在线脱气装置、自动进样器), Agilent OpenLAB CDS Chemstation(Edition C.01.07)网络工作站(Agilent科技有限公司)。安捷伦708-DS全自动溶出仪(配有850-DS自动取样器)。ES-E120B Ⅱ电子分析天平(天津市德安特传感技术有限公司);超声波清洗机(深圳市洁盟清洗设备有限公司)。
磷酸(质量分数为85%,色谱纯,成都市科龙化工试剂厂);甲醇、乙腈(色谱纯,山东禹王和天下新材料有限公司);娃哈哈纯净水(沈阳娃哈哈启力食品有限公司);庚烷磺酸钠(色谱纯,山东省禹城市中美色谱产品厂出品);吗啡对照品(批号171201-200822)、磷酸可待因(methylmorphine, MMP)对照品(批号171203-200504)和甘草苷(liquiritin, LQN)对照品(批号111610-200604)购自中国药品生物制品检定研究所;甘草酸铵(glycyrrhizic acid ammonium, CHAA)对照品(批号110731-201619)和苯甲酸钠(sodium benzoate, SB)对照品(批号100433-200301)购自中国食品药品检定研究院;芹糖甘草苷(liquiritin apioside, LQA)对照品(批号160524)、芹糖异甘草苷(isoliquiritin apioside, ILQA)对照品(批号170626)、甘草素(liquiritigenin, LQGN)对照品(批号160607)、异甘草素(isoliquiritigenin, ILQGN)对照品(批号160531)、异甘草苷(isoliquiritin, ILQN)对照品(批号170623)和新异甘草苷(neoisoliquiritin, NILQN)对照品(批号170720)购自上海融禾医药科技有限公司。
复方甘草片共145批,由厂家A(S1~S49)、厂家B(S50~S61)、厂家C(S62~S73)、厂家D(S74~S85)、厂家E(S86~S97)、厂家F(S98~S109)、厂家G(S110~S121)、厂家H(S122~S133)和厂家I(S134~S145)生产。
2.2 溶液的制备
双标溶液 分别取MP和CHAA对照品适量,精密称定,加甲醇制成200 mg/L MP和200 mg/L CHAA双标混合对照品溶液,摇匀,即得。
混合对照品溶液 分别取MP、LQN、MMP、SB和CHAA对照品适量,精密称定,加甲醇制成35 mg/L MP、60 mg/L LQN、16 mg/L MMP、160 mg/L SB、和800 mg/L CHAA的混合对照品溶液,摇匀,即得。
供试品溶液 取复方甘草片10片,研细,精密称取4片量,置于50 mL量瓶中,精密加提取溶剂(80%(v/v)甲醇溶液,含0.5%(v/v)磷酸)50 mL,精密称定,45 ℃超声处理(功率240 W,频率40 kHz)10 min,静置至室温,再精密称定,用提取溶剂补足减失的重量,摇匀,过0.45 μm滤膜,取续滤液,即得。
2.3 色谱条件
色谱柱为COSMOSIL 5C18-MS-Ⅱ柱(250 mm×4.6 mm, 5 μm);以0.2%(v/v)磷酸水溶液(含0.005 mol/L庚烷磺酸钠)为水相A,乙腈-甲醇(9∶1, v/v)溶液为有机相B,梯度洗脱,洗脱程序为0~10 min, 96%A~79%A; 10~20 min, 79%A~65%A; 20~32 min, 65%A~47%A; 32~45 min, 47%A~18%A; 45~50 min, 18%A~15%A; 50~55 min, 15%A~96%A;检测波长为220 nm,柱温为35 ℃,流速为1.0 mL/min。
3 结果与讨论
3.1 色谱条件的优化
以峰体相对信息指数Inrt(见公式(5))为优化目标函数对实验条件进行优化选择,峰数越多、均化性越好、柱效越高,则Inrt值越大,表明实验条件越理想。
(5)
其中pi、ri、Ai、Si、Ni、Λi分别是第i峰的面积归一化值、相关系数、峰面积、信息熵、理论塔板数和峰体指数,Ar、Λr为对照指纹峰面积和对照指纹峰体指数。
试验中根据以往经验确定一色谱条件,在此条件的基础上逐一对不同流动相组成(①0.2%(v/v)磷酸水溶液为水相,乙腈-甲醇(9∶1, v/v)为有机相;②0.2%(v/v)磷酸水溶液为水相,纯乙腈为有机相;③0.2%(v/v)磷酸水溶液(含0.005 mol/L庚烷磺酸钠)为水相,乙腈-甲醇(9∶1, v/v)为有机相),不同流动相梯度,不同提取溶剂(甲醇、50%(v/v)甲醇、80%(v/v)甲醇、80%(v/v)甲醇(含0.5%(v/v)磷酸))和不同厂家的C18色谱柱进行考察。将各色谱图积分后导入孙国祥等研发的“中药色谱指纹图谱超信息特征数字化评价系统4.0”软件并计算出Inrt值。结果表明,当采用2.3节色谱条件时,提取溶剂为80%(v/v)甲醇溶液(含0.5%(v/v)磷酸)时Inrt值最大。因此本实验确定最终以80%(v/v)甲醇(含0.5%(v/v)磷酸)作为提取溶剂,分离条件见2.3节。
3.2 方法学考察
3.2.1系统适用性试验
分别取适量MP、MMP、LQN、CHAA、SB、LQA、ILQA、LQGN、ILQGN、ILQN和NILQN对照品加甲醇充分溶解混匀制成各对照品溶液,另取供试品溶液(S1)适量,分别进样测定,见图1。对比保留时间和在线紫外光谱可知各对照品出峰位置,其中MP、LQN、MMP、SB、CHAA为复方甘草片指纹谱中第10号峰、14号峰、15号峰、16号峰和27号峰。因CHA峰与相邻色谱峰分离度高且峰形良好,因此选作参照物峰。另空针运行2 h,进样提取溶剂和供试品溶液运行2 h结果表明,在38 min后出现系统自身梯度峰,而样品在35 min前出峰基本结束,因此色谱系统对指纹图谱测定不产生干扰峰。考虑到梯度洗脱中色谱峰宽被显著压缩和保留时间被显著延长而导致梯度洗脱的塔板数值出现异常,孙国祥教授提出用压缩因子τ的平方校正理论塔板数,见公式(6),其中n为校正后的理论塔板数,tR为甘草酸峰保留时间,W1/2为甘草酸峰的半峰宽。其中CHA的τ的计算方法为:把梯度洗脱程序中初始有机相比例(4%)到目标物保留时间(CHA的tR=32.498)对应梯度段(第4段)段前的所有有机相比例累计值(即4+21+35+53=113)除以梯度段数,再除以初始有机相比例(从0洗脱时,除以第一段梯度速度),即τ2=(113/42)2=49.9。按甘草酸峰计算系统理论板数不应低于8 500,否则表明色谱系统不适用于测定复方甘草片的质量。
(6)

图 1 样品与对照品定位HPLC图Fig. 1 Identification chromatograms of sample and standards MP: morphine; LQA: liquiritin apioside; LQN: liquiritin; MMP: methylmorphine; SB: sodium benzoate: ILQA: isoliquiritin apioside; ILQN: isoliquiritin; NILQN: neoisoliquiritin; LQGN: liquiritigenin; ILQGN: isoliquiritigenin; CHAA: glycyrrhizic acid ammonium.
3.2.2精密度试验
精密吸取同一S1供试品溶液5 μL,连续进样测定6次,记录色谱图,以CHA的保留时间和峰面积为参照,各共有指纹峰相对保留时间的相对标准偏差(RSD)均小于1.0%,相对峰面积RSD均小于3.0%,表明检测系统进样精密度很好。
3.2.3稳定性试验
精密吸取同一S1供试品溶液5 μL,分别在溶液制备后0、2、4、6、14和22 h进样测定,记录色谱图,以甘草酸峰保留时间和峰面积为参照,各共有指纹峰相对保留时间的RSD均小于1.0%,相对峰面积的RSD除2号峰(RSD=6.0%)和25号峰(RSD=3.3%)外,其余均小于3.0%,表明样品在22 h内基本稳定。
3.2.4耐用性试验
分别在30、35、40 ℃柱温条件下进行试验,取S1供试品溶液进样5 μL,记录色谱图,以35 ℃下指纹图谱为标准(35 ℃下指纹图谱Sm=1.000,Pm=100%,)评价30 ℃和40 ℃下测得HPLC指纹图谱的宏定性相似度Sm分别为0.987和0.959,宏定量相似度Pm分别为99.7%和95.8%,随着柱温升高,色谱峰保留时间缩短,但指纹峰数量无差异,各指纹峰大小有显著差异;此外,分别在0.8、0.9、1.0、1.1、1.2 mL/min流速时,取S1供试品溶液进样5 μL,记录色谱图。由谱图可知流速降低时各指纹峰保留时间延后,各指纹峰面积加大,从而导致宏定量相似度明显增大,流速增大时宏定量相似度降低,Pm与流速(v)呈线性关系,Pm=-102v+202.26,R2=0.996 0。此时系统分离情况差别不大,但宏定量相似度发生显著变化,流速为1.0 mL/min时各指纹峰保留时间适中且峰形最好。因此,根据定量指纹图谱特殊性需要,保持流速1.0 mL/min和柱温35 ℃。
3.2.5方法重复性试验
按供试品溶液制备方法制备S1样品供试液6份,分别精密吸取5 μL进样测定,每个供试液进样测定2次,记录色谱图,以甘草酸峰为参照物峰确定28个共有指纹峰,以平均值法生成标准指纹图谱,用该标准指纹图谱为对照标准由软件计算评价12次测定的指纹图谱相似度结果,同一样品二次结果取平均。结果6份供试品指纹图谱的宏定性相似度均值为0.999, RSD=0.3%(n=12);平均宏定量相似度为100.0%, RSD=0.6%(n=12),另根据5个Q-markers的峰面积计算出含量,可知MP、LQN、MMP、SB和CHAA的含量RSD均小于3.0%,表明方法重复性很好。
3.3 复方甘草片指纹图谱的建立和评价
按2.3节色谱条件测定9个厂家共145批复方甘草片,记录色谱图。将积分后*. CDF文件导入孙国祥等研发的“中药色谱指纹图谱超信息特征数字化评价系统4.0”软件,以甘草酸峰保留时间和峰面积为参照,确定28个共有指纹峰,按平均值法生成标准指纹图谱(RFP)。由于样品包含了中国复方甘草片主要生产厂家样品,其代表了整个行业产品质量的基本特征,所得到的RFP可作为复方甘草片标准制剂的标准指纹图谱(SP-RFP)。以此SP-RFP为评价标准,供试品指纹图谱与RFP的宏定性相似度不得低于0.90,宏定量相似度应在80%~120%。以此标准评价145批次复方甘草片28个主组分指纹的整体量值结果。以Sm和Pm为参数进行聚类分析,采用SPSS软件进行系统聚类,结果表明S75、S81~S85、S107~S109为第Ⅰ类,其余为第Ⅱ类。虽然Ⅰ类Pm数值偏大(112%~117%之间),但全部在质量规定范围内,因此不剔除任何批次样品。9个厂家的样品评价结果见表1。根据9个厂家的样品Pm绘制的生长曲线见图2。可看出厂家D的样品整体Pm最大,厂家A的样品整体Pm最小,厂家C的样品Pm波动最小,厂家F的样品Pm值波动最大。以上9厂家样品均合格,其中厂家A生产批号为20171213、厂家B生产批号为PDE0712、厂家E生产批号为3180502、厂家G生产批号为20180620以及厂家H生产批号为18053001和18060501的复方甘草片的Sm≥0.95,Pm≈100%,可初步选作复方甘草片的标准制剂。试验中每隔一段时间进样测定双标溶液,记录色谱图,计算相对定量校正因子,考察色谱系统是否发生变化,结果见表2。结果表明在复方甘草片特征指纹图谱研究期间以及样品测定期间,系统定量性质无变化(fqi在0.97~1.03之间),不需对色谱系统进行双标校正。

表 1 系统指纹定量法评价9个厂家复方甘草片质量的一致性与5个Q-markers的含量结果Table 1 Quality consistency results for Fufanggancao tablets (FFGCTs) produced by nine manufactures evaluated by the systematic quantitative fingerprint method and the contents of the five Q-markers
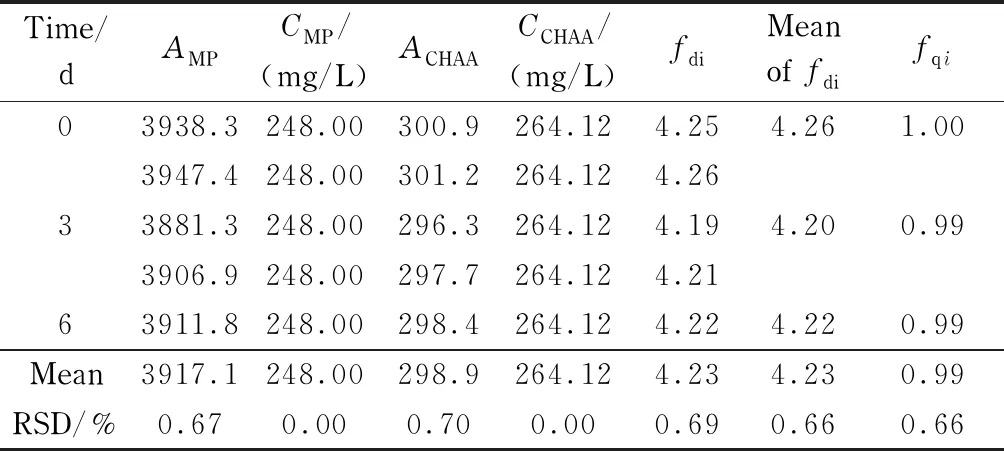
表 2 复方甘草片定量指纹图谱的双标校正因子Table 2 Double marker calibration factors of Fufanggancao tablets quantitative fingerprints
A: peak area;C: mass concentration, mg/L;fdi: the absolute quantitative correction factor;fqi: the relative quantitative correction factor.
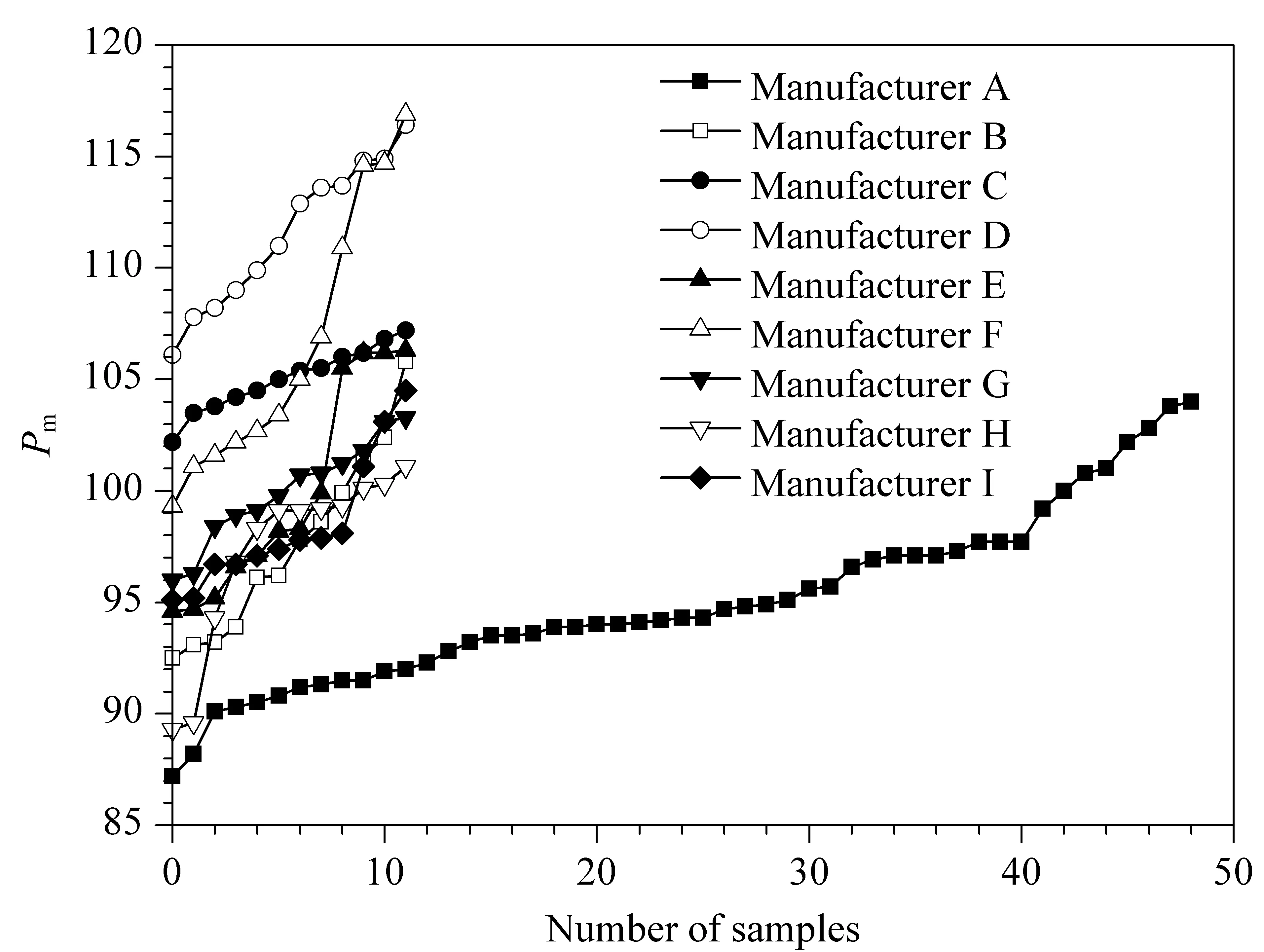
图 2 9个厂家复方甘草片的Pm生长曲线图Fig. 2 Pm growth curves of FFGCTs from nine manufacturers
3.4 含量测定法
3.4.1线性关系考察
精密称定MP、LQN、MMP、SB和CHAA适量,用甲醇稀释,制成6个质量浓度的混合对照品溶液,每份溶液在上述色谱条件下各平行测定两次,以峰面积均值(Aavg)对各对照品浓度(C, mg/L)进行回归,结果见表3。5组分在各线性范围内的线性关系均很好。以信噪比10∶1为定量限,信噪比3∶1为检出限。取MP、LQN、MMP、SB和CHAA对照品适量,加甲醇溶解并稀释制成一定浓度的溶液,进样检测。测得MP、LQN、MMP、SB和CHAA的定量限与检出限如表3。

表 3 5个Q-markers的线性方程、线性范围、定量限和检出限Table 3 Linear regression equations, correlation coefficients, linear ranges, LOQs and LODs of the five Q-markers
A: peak area;C: mass concentration, mg/L.
3.4.2精密度和稳定性试验
以混合对照品溶液中5种组分峰面积的RSD评价仪器精密度和供试液的稳定性,结果RSD均小于3.0%,表明仪器精密度和供试液稳定性均满足定量分析要求。
3.4.3加样回收试验
分别取S1样品6份,分别精确加入MP、LQN、MMP、SB和CHAA对照品适量,按2.2节方法处理,进样测定。另按照2.2节下方法配制供试品溶液与混合对照品溶液,进样测定。以峰面积计算回收率。结果5种组分的平均回收率分别为100.3%、98.9%、103.0%、98.9%和99.4%,其RSD均不大于1.0% (n=6),表明方法准确度很好。
3.4.4样品含量测定
将145批复方甘草片供试品溶液进样测定,用标准曲线计算供试品溶液中MP、LQN、MMP、SB和CHA的含量,其中计算CHA含量时应在CHAA标准曲线计算所得值基础上乘以0.979 7[17]。9个厂家的样品中5个Q-markers的含量见表1。厂家D的样品中吗啡和苯甲酸钠含量均高;厂家B的样品中甘草苷和甘草酸铵平均含量均最高;而各厂家样品中磷酸可待因的平均含量差异不大。《中国药典》规定每片复方甘草片中吗啡含量应为0.36~0.44 mg,甘草酸含量不得少于7.3 mg[17],除厂家G生产的复方甘草片中吗啡平均含量没有达到要求外,其他厂家生产的复方甘草片中吗啡和甘草酸含量均达到药典要求。以上9个厂家样品中各组分含量差异可能是源于所用原料药不同或者工艺不同。
3.5 原料指纹与制剂的相关性及质量的智能预测
3.5.1复方甘草片指纹归属与指纹分解亚定量相似度

(7)
其中,xi为样品图谱中各指纹峰的峰面积,yi为对照图谱中各指纹峰的峰面积。
3.5.2制剂与甘草浸膏粉标准指纹图谱的相关度
用统一化色谱条件测定70批甘草浸膏粉指纹图谱,并获得甘草浸膏粉的RFP,以复方甘草片的SP-RFP为标准,用实验室研发的软件剔除罂粟果提取物粉和苯甲酸钠的指纹峰后,定量评价甘草浸膏粉RFP,结果Sm=0.988,Pm=98.5%,由此可看出二者宏定量相似度误差为1.5%,在测定误差范围内。因此,可以用复方甘草片SP-RFP评价原料药物甘草浸膏粉的质量,评价时只需从评价组峰里删除罂粟果提取物粉和苯甲酸钠的4个指纹峰(所得评价结果的Sm需要除以0.988,Pm除以98.5),以达到智能预测甘草浸膏粉质量的作用,避免劣质原料入药。相反,也可用3.5.1节下配制模拟样的方法简单预测所用原料药制得的制剂质量是否合格,避免生产出不合格的药品。
3.6 复方甘草片紫外全指纹溶出度测定法
紫外全指纹溶出度测定法,依据测定的190~400 nm紫外光谱的211个指纹点来监测中药固体制剂中整体主组分的溶出度[16,19,20]。实验中测定了厂家A、厂家B、厂家F、厂家G以及厂家I等5个厂家复方甘草片溶出曲线,每个厂家各测定3批。分别以水(2 h在线紫外溶出光谱作为全溶出UV-RFP评价标准)和0.1 mol/L盐酸作为溶出介质(5 h在线紫外溶出光谱作为全溶出UV-RFP评价标准),以最终时间点的图谱为评价标准,将测得的不同时间点的图谱经软件评价后测得的Pm值作为样品的溶出度,3批样品取平均值后绘制平均溶出度曲线,5个厂家的样品在以水为介质和以0.1 mol/L盐酸为介质时溶出曲线趋势大致相同,因此以水介质溶出曲线为例,见图4。从结果可看出,5个厂家生产的复方甘草片溶出情况存在显著差异,其中以水和0.1 mol/L盐酸作为溶出介质时厂家F生产的复方甘草片溶出速度均最快。此法可以用于评价复方甘草片制剂工艺的合理性。规定在水中30 min时溶出度不得低于40%;在0.1 mol/L盐酸溶出介质中30 min时的溶出度不得低于30%。采用900 mL溶出介质和每次取液2.0 mL,实验中废弃液体的体积为0.5毫升,转速50 r/min。

图 4 5个厂家的复方甘草片在水介质中的平均溶出度曲线Fig. 4 Average dissolution curves of FFGCTs from five manufacturers obtained in water
4 结论
本文是对复杂样品体系的质量一致性评价的方法模式探讨和方法步骤探讨。采用大数据法建立了复方甘草片标准制剂HPLC定量指纹图谱,采用系统指纹定量法对28个主组分指纹进行整体定性和整体定量一致性控制。同时对MP、LQN、MMP、SB和CHA进行精准定量控制,实现对复方甘草片质量的一致性评价。此外本文构成原料药与制剂的统一化色谱系统,可对制剂指纹峰进行来源归属,也可实现对原料药物与制剂质量的智能预测,以防止低劣原料入药,另结合紫外全指纹溶出度评价制剂工艺的合理性,在生产实践中具有重要意义。这为复方甘草片质量一致性评价提供了全面、可靠的评价方法。复方甘草片为目前我国一致性评价中289个品种之一,属于特殊品种。孙国祥课题组自2003年开始一直立足于定量指纹图谱理论和方法研究,以及中药标准制剂的概念研究,打破传统思路,同时又密切结合中药质量控制特点和《中国药典》质量控制方法,形成了系统、规范的中药和植物药质量一致性评价方法,为未来我国中药和天然药物进行质量一致性评价和制剂工艺一致性评价提供了重要参考先例。
