单原子催化CO2加氢还原-DFT研究
2019-10-08苏学冰周俊杰
苏学冰,周俊杰
(郑州大学化工与能源学院,河南 郑州 450001)
全球每年CO2排放量超过300亿t,因此,急需研究一种绿色环保的利用方法将CO2转化为具有价值的化学产品和燃料[1,2]。 将CO2捕集、封存和利用是目前减少并转化CO2的主要策略[3],目前已经有多种方法将CO2转化利用,如化学转化、光化学转化以及电化学方法。 众所周知,CO2化学形式相对惰性,很难提高其转化选择性及其活性。 逆水煤气变换反应(RWSG) 是将CO2转化为CO资源重要的化学利用方法[4],中间体CO可以继续加氢合成其他化学产品。陶旭梅等[5]通过量子化学方法研究CO2与氢气合成甲醇转化路径,发现最优转化路径为CO加氢机理。 有研究表明RWSG反应在Ni、Rh以及Cu金属表面首先进行CO2解离,而在Pt、Ag以及Pd金属表面首先加氢得到羧基[6]。
负载金属催化剂已在工业上实现商业化应用,相对于金属负载催化剂,亚纳米团簇催化剂显示更高的催化活性[7],主要是由于亚纳米团簇使得更多的过渡金属暴露在表面,与反应物有更多的接触表面。近年来,单原子催化(SAC)的研究已应用于多种反应中,如一氧化碳氧化、二氧化碳还原以及甲烷重整制氢等领域[8],具有较高的活性位点以及高原子利用率。 马丁等[9]研究金属Ir粒径与CO2加氢还原产物(如CO)的关系,研究表明Ir粒径越小与金属载体的结合能力越强,并其高选择性地生成CO,抑制了高温条件下CO2加氢还原生成甲烷。 Seoin等[10]采用密度泛函方法将不同金属原子负载在石墨烯上研究单原子催化剂对CO2电化学转化利用的影响。Wu Ping等[11]采用密度泛函方法用石墨烯负载金属Fe研究CO氧化反应,研究表明石墨烯是很好的金属负载物,能阻止Fe的团聚作用并降低CO氧化的活化能。 He等[12]利用石墨烯为负载物,采用第一性原理研究金属Ag、Cu、Pd、Pt以及Co上CO2加氢还原合成CH4和CH3OH转化反应路径。 研究表明在单金属Cu上CO2易合成CH3OH。Cheng等[8]N掺杂改性石墨烯合成金属Co-N-C在高温下具有较高的稳定性。
综上所述, 金属Cu对CO2加氢还原具有催化作用, 石墨烯是很好的单金属催化剂负载物且N掺杂的石墨烯的载体在高温下具有更高的稳定性。 因此本文采用密度泛函方法,建立Cu-N-C催化剂模型和Cu(111)表面模型研究N掺杂石墨烯负载金属铜催化剂对CO2加氢与解离影响。
1 计算模型和方法
建立单层周期性石墨烯晶体模型,模型中包含24个碳原子并在z方向建立1.5nm的真空层, 如图1a所示,建立Cu-N-C模型,将其中的4个碳原子替换为3个N原子和一个Cu金属原子。 建立Cu表面模型选取3层平板厚度和3×3超晶胞模型,来模拟Cu(111)表面,相邻两层平板的真空层厚度为1nm,以确保平板间的相互作用足够小。 Cu(111)表面模型有4个吸附位,分别为top、bridge、fcc和hcp位。 如图1b所示。
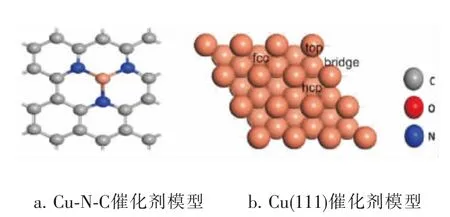
图1 催化剂模型
计算采用交换相关势,采用广义梯度积分(GGA)和Perdew-Wang-91泛函相结合的方法(GGA-PW91),金属原子采用ECP赝势; 价电子波函数采用双数值基组加极化函数(DNP)展开,能量、受力和位移收敛标准分别为2×10-5Ha、4×10-3Ha/10-10m和5×10-3×10-10m,布里渊区积分的Monkhorst-Pack网格参数设置为2×2×1,拖尾效应(smearing)设置为0.005Ha。 过渡态计算使用Complete LST/QST完全线性同步/二次线性同步方法。 对于反应的吸附能,通常是指吸附物质与底物之间的相互作用力,其符号大小可以表示发生吸附的可能性和吸附程度。对于物质A来说,其吸附能Ead定义为:

式中:EA/slab表示物质吸附在底物上的总能量;Eslab表示底物能量;EA表示自由吸附质A的能量。Ead越大其吸附作用力就越强。
2 结果与讨论
2.1 计算模型以及方法验证
为了验证计算方法的正确性,首先优化纯净石墨烯模型,计算得到石墨烯中C-C键为1.423×10-10m,与文献值1.420×10-10m相近[13],为验证负载金属模型的合理性,建立石墨烯负载Cu单原子模型,计算Cu-C模型中得到Cu-C键键长为1.907×10-10m、1.878×10-10m、1.864×10-10m,与文献值1.88×10-10m相吻合[12],计算铜晶胞的晶格参数α=3.589×10-10m与文献值[14]α=3.615×10-10m误差仅有0.72%。 说明计算所采取的模型方法可以应用于Cu-N-C催化剂模型中。
2.2 CO2、COOH在催化剂表面吸附
CO2分子及其加氢产物COOH在催化剂Cu(111)面与Cu-N-C催化剂表面吸附构型及其结合能如表1所示。

表1 CO2与COOH在催化剂表吸附能及其结构参数
从吸附构型看,在催化剂Cu-N-C上,优化后CO2的键角θO-C-O为151.2°,而在催化剂Cu(111)面CO2基本不变形,可以看出Cu-N-C对CO2的作用力更大。 在结合能方面,CO2在催化剂Cu-N-C上结合能远远大于其在Cu(111)催化剂表面的结合能,且属于典型的化学吸附,而在Cu(111)面属于典型的物理吸附。
2.3 CO2加氢活化
CO2加氢主要得到COOH和HCOO, 因为HCOO在催化剂表面相对于CO2加氢还原中其他中间产物极其稳定,一般不考虑HCOO[15]。 本文主要考虑仅CO2第一步加氢还原生成COOH物种的反应。
搜索反应CO2+H→COOH反应过渡态, 在催化剂Cu(111)上,H原子向CO2中的一个氧原子靠近,同时CO2分子向催化剂表面迁移,H-O键长为0.146nm,其中C原子与另一个O原子在top位与Cu形成化学键。 此过程反应活化能垒为172.34kJ/mol,反应前后能差为-53.13kJ/mol,为放热反应。
在催化剂Cu-N-C上,H首先吸附在其中的一个碳原子上,CO2吸附在Cu原子上, 反应过程中H向CO2方向迁移并靠近其中的一个O原子,CO2同时向H方向移动,最终形成COOH,此过程所需反应活化能为55.06kJ/mol,反应前后能差为-50.58kJ/mol,反应为放热反应,如图2所示,为CO2在不同催化剂上加氢合成COOH物种的势能图。 从反应势能如可以发现,CO2在催化剂Cu-N-C上合成COOH所需的反应活化能远小于CO2在Cu(111)面反应所需的活化能。 说明单原子催化剂Cu-N-C对CO2加氢活性更高。
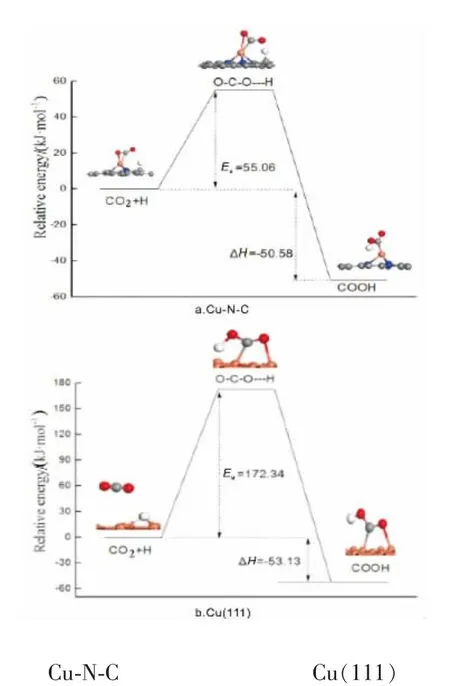
图2 CO2 加氢反应势能图
COOH可以进一步分解得到CO分子,CO是CO2加氢还原过程中重要的反应物质。 对COOH分解得到CO和OH反应进行过渡态搜索,在催化剂Cu(111)面,CO 与OH 之 间 的C-O 键 长 由0.135nm 拉 长 至0.172nm,最后C-O键断裂形成CO和OH,最终CO迁移至临近的桥位,OH迁移至临近的fcc位;该过程反应活化能垒为35.77kJ/mol, 反应前后能差为-53.33kJ/mol,为放热反应,反应势能图如图3所示。
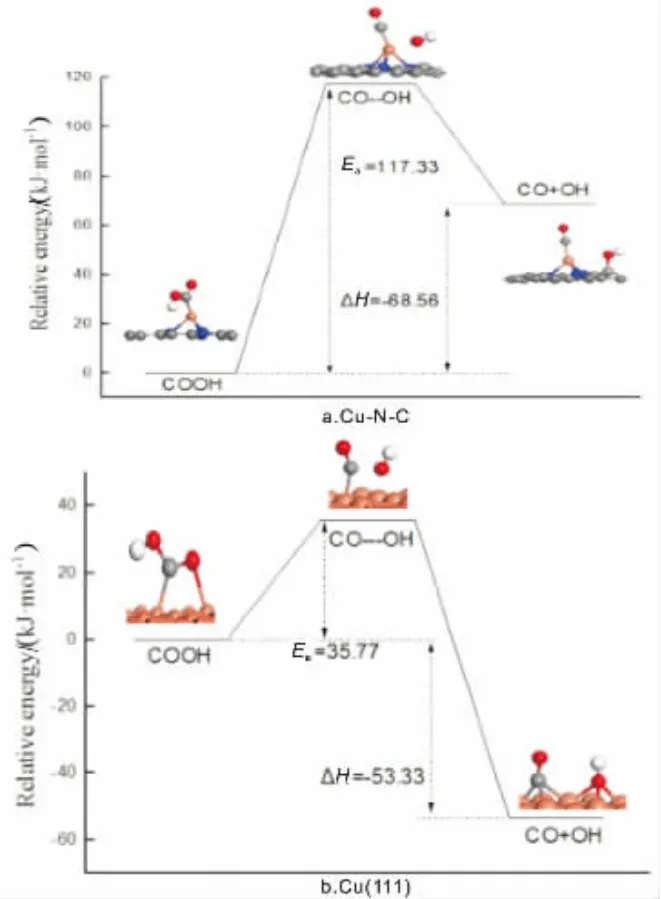
图3 COOH 解离势能图
在催化剂Cu-N-C上, 对于O-O键断裂,CO稳定地吸附在Cu原子上, 而OH吸附在C以O-C键与石墨烯表面碳原子结合。 此过程所需反应活化能为117.33kJ/mol,反应前后能差为68.56kJ/mol,反应为吸热反应;从势能图3上可以看出,相对于Cu(111),催化剂Cu-N-C对COOH的分解作用不大。 从整个反应过程看,CO2加氢在催化剂Cu-N-C所需的最大反应活化能相对于催化剂Cu(111)的小,N掺杂石墨烯负载的Cu催化剂对CO2反应活性更高。
CO2可以在催化剂表面分解成CO和O物种,如图4所示。搜索CO2在Cu(111)面分解过程发现,该过程活化能垒Ea为153.09kJ/mol, 反应前后能差为90.94kJ/mol,为吸热反应。在Cu-N-C上,CO以Cu-C键形式作用于Cu原子,O移动至C原子上,分析该过程所需活化能垒为342.48kJ/mol, 反应前后能差为212.47kJ/mol,说明在Cu-N-C上CO2不易分解CO和O物种。
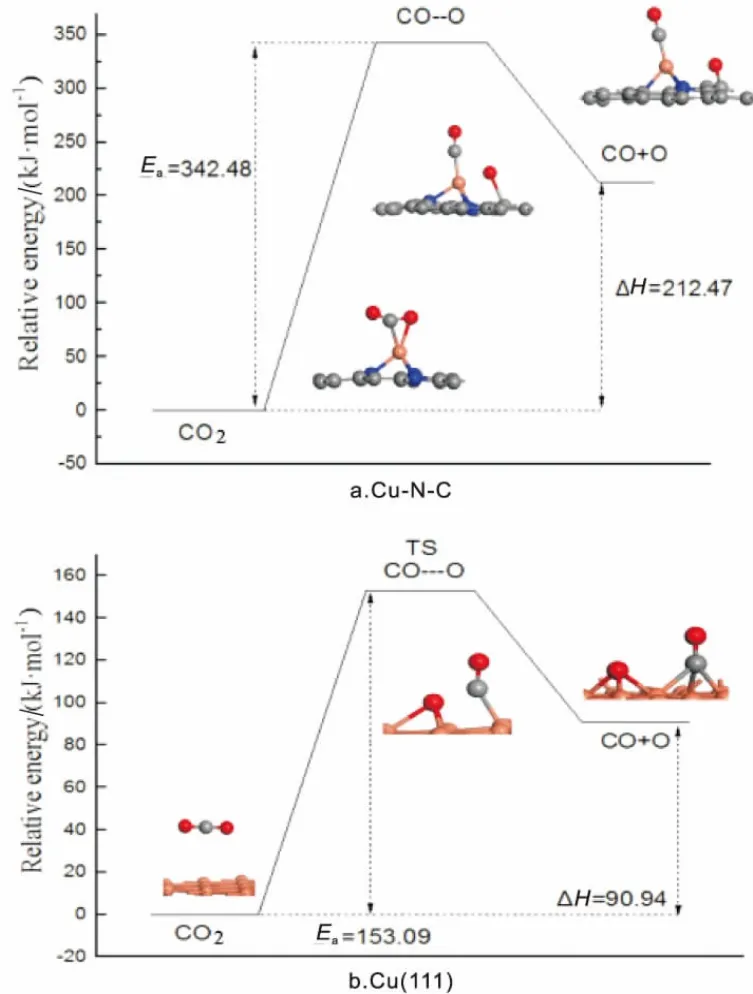
图4 CO2解离势能图
通过对比分析CO2在Cu(111)与Cu-N-C上各基元反应活化能得到,在Cu(111)面,相比CO2加氢生成COOH,CO2易于直接分解成CO和O这一结果与文献结果一致[6]。 而在Cu-N-C上,CO2易于先加氢生成COOH,通过COOH分解得到CO和OH。
2.4 催化剂电子结构
为了分析Cu-N-C电子结构性质以及更好解释Cu-N-C催化作用,因此研究Cu-N-C中Cu、N以及C的偏态密度图(PDOS),并研究了CO2在催化剂表面吸附电子特性。
图5为催化剂Cu-N-C的偏态密度图 (PDOS)和表面电荷以及LUMO分布图。由图中可以看出,Cu的d轨道与N的p轨道发生杂化,N的s轨道与C的s轨道在0.75eV峰值相重合发生较强杂化。而N与C的2p轨道基本重合,说明N与C发生强相互作用。 由表面电荷分布图可知,Cu将电荷传递给基底形成正电位易于接受电子。 同时分析Cu-N-C的最低空轨道分布可知,LUMO主要集中在Cu原子上, 说明Cu是易于进行反应的位点。
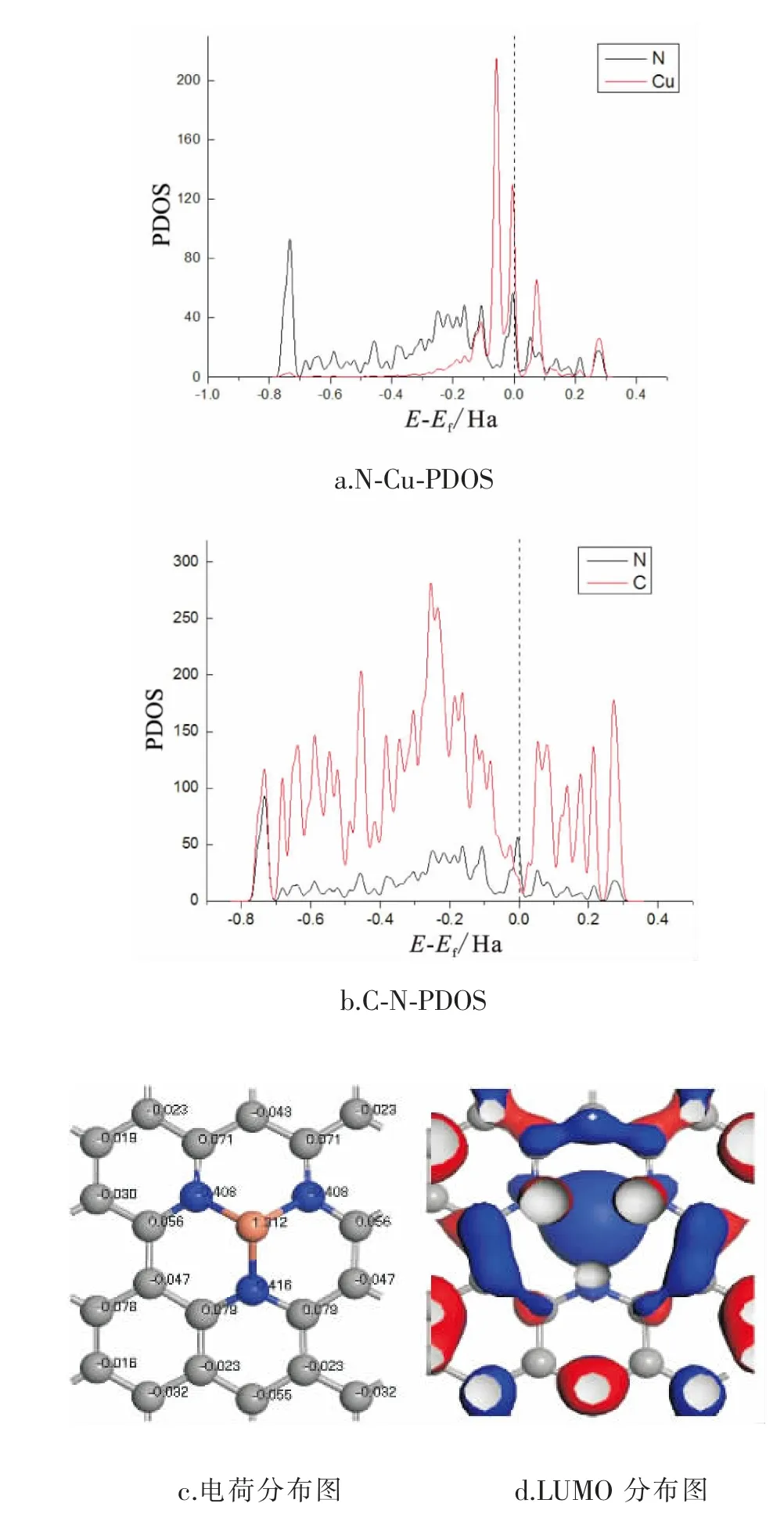
图5 催化剂表面电子结构

图6 CO2吸附在催化剂表面PDOS图
图6 为CO2吸附在催化剂Cu-N-C与Cu(111)中吸附分子CO2与Cu的偏态密度 (PDOS), 由图可以看出,吸附态CO2中的C原子与O原子的p轨道与Cu-N-C催化剂Cu的d轨道有较为明显的重合,而与Cu(111)中Cu原子的d轨道重合较小, 说明CO2与催化剂Cu-N-C的d轨道发生较强的杂化, 具有较强的相互作用,而与Cu(111)相互作用不大,这与前面计算所得到的结合能结果一致。
3 结论
通过密度泛函理论研究了N改性的石墨烯负载单原子Cu催化剂对CO2加氢还原的作用, 并利用电子态密度分析了催化剂Cu-N-C与Cu(111)电子结构性质和CO2吸附态密度图。 研究结果表明,在单原子催化剂Cu-N-C对CO2的吸附作用相比纯金属Cu的吸附作用更强,Cu-N-C中Cu原子d轨道与CO2中的p轨道发生较强杂化;CO2在Cu(111)面上更倾向于直接分解得到CO, 而在Cu-N-C上,CO2易于加氢生成COOH分解得到CO,且整个反应所需活化能能小于Cu(111)面上所需活化能。 与Cu(111)催化剂相比,单原子催化剂Cu-N-C降低CO2加氢合成COOH所需活化能,升高COOH分解所需活化能,但就CO2加氢还原生成CO过程相比,降低过程中关键反应所需的活化能,对RWSG反应具有更高的催化效果。分析催化剂Cu-N-C电子结构得到Cu的d轨道与N改性的石墨烯的p轨道有很强的相互作用,结构稳定性好。
