牛妊娠相关糖蛋白1(bPAG1)的真核表达及纯化
2019-10-08刘长彬卢春霞张云生石国庆1
刘长彬,卢春霞,杨 华,张云生,石国庆1,*
(1.石河子大学 动物科技学院,新疆 石河子 832003; 2.新疆农垦科学院 省部共建绵羊遗传改良与健康养殖国家重点实验室,新疆 石河子 832000;3.长江师范学院 现代农业与生物工程学院,重庆 408100)
【研究意义】妊娠相关糖蛋白 (Pregnancy-associated glycoproteins,PAG) 是偶蹄类动物胎儿胎盘滋养层细胞合成、分泌的一类糖蛋白。1991年Zoli 等[1]从奶牛胎盘中分离得到分子量为67 kDa的糖蛋白,等电点为4.4、4.6、5.2和5.4,与Butler等[2]发现的妊娠特异性蛋白 B (PSPB)有着高度的相似性,构成了一个庞大的胎盘糖蛋白家族,统称PAG。PAG属于天冬氨酸蛋白酶家族,与胃蛋白酶、组织蛋白酶D、组织蛋白酶E有50 %以上的相同氨基酸序列,但大多数PAG由于催化位点氨基酸发生置换使其没有酶活性。目前,牛妊娠相关糖蛋白(bPAG)基因家族至少有 22 个转录本以及变异体,根据其出现的时间,将其分为两组:“古代组”与“现代组”,大多数 PAG属于“现代组”,由胎盘滋养外胚层的双核细胞表达产生,后经双核细胞迁移而进入血液,而“古代组”PAG 则在整个滋养外胚层细胞中产生[3-5]。另外,不同bPAG在妊娠期内的表达水平和表达时间有明显区别[5-7]。【前人研究进展】在bPAG家族中,bPAG1在母畜怀孕后不久就能外周血中检测到,并在整个孕期持续存在,常作为一种标志物用于家畜早期妊娠诊断[8-10]。目前,PAGs 商业化检测试剂盒在国外已经得到广泛的应用,并取得较高的准确率[11-12],但高的检测成本(每头次约33元)使其在国内无法大规模推广应用。而国内关于PAG的研究报道极少,且无商品化的PAG抗原销售,进而限制了PAG快速检测方法的研发及应用。因此,如何获取高纯度的PAG蛋白,是制备PAG抗体和研发快速检测产品的基础,是解决生产中实际问题的关键环节。【本研究切入点】目前,前人多采用磷酸钾缓冲液提取、饱和硫酸铵沉淀、离子交换层析等传统方法从母畜胎盘子叶中分离纯化天然PAG蛋白[1,13-14],但步骤繁琐复杂,且在纯化过程中稍有不慎容易导致蛋白降解。虽然有研究者利用豌豆、扁豆、紫藤花凝集素[15-17]以及胃蛋白酶抑制剂[18-19]特异性结合PAG,通过亲和层析法获得PAG,但该法成本较高,不利于PAG的大量纯化。而体外重组蛋白技术为PAG结构、功能和应用研究提供了新思路[20-21]。目前,国内关于bPAG1重组蛋白的研究鲜有报道。【拟解决的关键问题】基于此,本研究对bPAG1密码子进行优化,通过全基因合成法得到bPAG1基因序列,然后构建PCDNA3.1 (-)-bPAG1真核表达载体,在中国仓鼠卵巢(Chinese hamster ovary, CHO)细胞中诱导表达并产生重组bPAG1,为奶牛早期妊娠诊断产品的研发奠定基础。
1 材料与方法
1.1 材料与试剂
CHO细胞株由本实验室保存;PCDNA3.1 (-)载体购自美国Invitrogen公司;TOP10感受态细胞、小鼠抗6× His单克隆抗体、DNA mark、TaqDNA 聚合酶、Pfu DNA 聚合酶、T4DNA 连接酶、EcoRI 内切酶、HindIII 内切酶、脂质体高效转染试剂、动物细胞蛋白裂解液、Ni-IDA 蛋白纯化试剂盒、DNA 琼脂糖凝胶回收试剂盒、SDS-PAGR变性丙烯酰胺凝胶快速制备试剂盒、柱式质粒DNA抽提试剂盒、BCA蛋白检测试剂盒等购自生工生物工程(上海)股份有限公司。PVDF膜购自美国Millipore公司;Expi293TMExpression Medium 购自上海北诺生物科技有限公司。
1.2 仪器与设备
VeritiTM96梯度PCR仪(美国ABI);Gel Doc XR+凝胶成像系统(美国BIO-RAD);Essential V4凝胶成像系统(英国UVITEC);Powerpac 300电泳仪(美国BIO-RAD);DYCZ-24EN双垂直电泳槽(北京六一仪器厂),DYCP-31BN琼脂糖水平电泳槽(北京六一仪器厂);Thermo311 CO2培养箱(美国热电公司);Thermo ScientificTMVarioskan Flash全波长扫描式多功能读数仪(美国赛默飞);转膜仪(美国Invitrogen公司);WD-9405A型脱色摇床(北京六一仪器厂);5424R台式冷冻离心机(德国eppendorf公司);BSC-150 型恒温恒湿培养箱(上海博讯事业有限公司);优普超纯水制造系统(成都超纯科技有限公司)。
1.3 方法
1.3.1 bPAG1基因优化与合成 根据Genbank公布的bPAG1(登录号:M73962)核苷酸序列,以及宿主细胞的对密码子的偏好性,对bPAG1密码子进行优化,并在优化序列的5’端设计EcoRI位点,3’端设计HindIII酶切位点和His-Flag 标签,优化的基因序列见3.1。根据优化后的bPAG1核苷酸序列,应用 primer premier软件设计54对引物(其余52对引物未列出),上游引物1:GACACGAATTCGCCACCATGCCCCTGCTTCTTTTGCT,下游引物54:GTGTCAAGCTTCTATTTATCGTC(下划线分别分为EcoRI 和HindIII酶切位点),然后采用全基因合成法获得目的产物,bPAG1全基因合成由生工生物工程(上海)股份有限公司完成。PCR扩增产物经1.0 %琼脂糖凝胶电泳检测, DNA 凝胶回收试剂盒回收目的片段。
1.3.2 重组载体PCDNA3.1(-)-bPAG1的构建 用EcoRI和HindIII限制性内切酶将回收的PCR产物和PCDNA3.1(-)载体进行双酶切。PCR产物酶切体系:纯化回收的片段1 μg (20 μl),10×FD Buffer 5 μl,EcoRI 1 μl (10 U/μl),HindIII 1 μl (10 U/μl),ddH2O加至50 μl。PCDNA3.1-载体酶切体系:PCDNA3.1(-)载体1 μg, 10× FD Buffer 5 μl,EcoRI 1 μl (10 U/μl),HindIII 1 μl (10 U/μl),ddH2O 加至50 μl,以上体系分别37 ℃酶切2 h。2种酶切产物经琼脂糖凝胶电泳检测后,用胶回收试剂盒回收。将回收的酶切产物用T4连接酶在22 ℃连接2 h。连接体系为:目的片段8 μl,酶切空载体4 μl, T4DNA Ligase 1 μl (5 U/μl),10×T4DNA Ligase buffer 2 μl,ddH2O 加至20 μl。
1.3.3 重组载体PCDNA3.1(-)-bPAG1的转化、筛选与鉴定 将10 μl连接产物加入到50 μl的TOP10F'感受态细胞中,混匀,冰浴 10 min。42 ℃热激 45 s,冰浴 5 min。加入 800 μl LB液体培养基中,37 ℃ 摇菌培养45 min。取20 μl菌液涂到Amp+/LB 固体培养基平板上,37 ℃培养 14 h。挑取单个菌落接种到Amp+/LB液体培养基中,37 ℃摇菌培养6 h,采用质粒抽提试剂盒提取质粒,以其为模板进行PCR鉴定。PCR反应体系:上游引物1 1 μl,下游引物54 1 μl,模板 3 μl,TaqDNA聚合酶 1 μl (10 U/μl),dNTP 1 μl, 10× PCR buffer 5 μl,ddH2O补齐至50 μl。PCR反应条件:95 ℃预变性3 min, 95 ℃变性22 s,58 ℃退火20 s,72 ℃延伸30 s,共30个循环,72 ℃延伸5 min,将PCR产物进行琼脂糖凝胶电泳检测。并对提取的质粒进行双酶切鉴定。将得到的阳性转化子送生工生物工程(上海)股份有限公司测序,经测序验证插入序列正确无误后,进行质粒大量抽提,保存至-20 ℃备用。
1.3.4 重组质粒PCDNA3.1(-)-bPAG1的诱导表达 rbPAG1的诱导表达参考Patel的方法[22],并稍作修改。将CHO细胞接种于含6 mM L-glutamine的Expi293TM培养基中,使细胞浓度达到106cells/mL,37 ℃、5 % CO2培养过夜。将100 μg重组质粒取和200 μl转染试剂加入至20 mL Expi293TM培养基中,涡旋混匀,室温孵育10 min。然后将混合液加入至上述CHO培养物中,37 ℃、5 % CO2培养4 d,收集表达上清,1000 r/min离心5 min,过0.22 μm滤膜。
1.3.5 重组蛋白的纯化 收集CHO培养液上清,采用Ni柱亲和层析法纯化目的蛋白。首先使用含10 mM的Binding buffer以1 mL/min的流速平衡Ni柱,将上清与Binding buffer混合,上样,用两倍柱体积的Binding buffer冲洗Ni柱直至紫外回复至基线处,除去杂蛋白,然后采用含不同浓度的咪唑(10、40、80、250、500 mM)洗脱溶液进行梯度洗脱,将上清液、流穿液、洗脱液进行 SDS-PAGE电泳,通过考马斯亮蓝染色检测纯化效果。同时采用BCA试剂盒测定蛋白浓度。
1.3.6 重组蛋白的Western blot鉴定 纯化后的rbPAG1经SDS-PAGE电泳后,取下凝胶,采用半干式电转印法将rbPAG1转至PVDF膜上。PVDF 膜使用之前在中甲醇中浸泡 30 s,之后将 PVDF 膜、凝胶、滤纸置于转膜液浸泡,浸泡后将三者按照正确顺序放在转印仪上,转印 45 min。取下 PVDF 膜于 5 %脱脂奶粉中,室温封闭2 h。用1×PBST漂洗3~4次,每次5 min。加入1∶2000 稀释的抗 His 标签小鼠单克隆抗体,室温孵育1 h,用1×PBST漂洗3~4次,每次5 min。最后用ECL发光底物,拍照。

图1 bPAG1基因PCR扩增电泳图Fig.1 The electropherogram of PCR products of bPAG1 gene
2 结果与分析
2.1 bPAG-1基因 PCR扩增与鉴定
Genbank(登录号:M73962)公布的bPAG-1基因全长为1295 bp,蛋白编码区(Coding sequence, CDS)长度为1143 bp,包含9个外显子和8个内含子[23],根据宿主细胞对密码子的偏好性,优化后的bPAG-1基因序列全长1218 bp,基因序列:GAATTCGCCACCATGCCCCTGC TTCTTTTGCT GCCACTGCTC TGGGCCGGGG CCCTGGCCAT CGTGAAGATT CCCCTGAGAA GACTTAAGAC AATGAGAAAT GTGGTGAGTG GGAAAAACAT GCTTAACAAC TTTCTCAAGG AGCACGCCTA CAGCCTGTCT CAAATTTCTT TCCGCGGGTC AAACCTGACA ACACATCCCC TGCGCAACAT CAAGGATCTG GTGTATATGG GGAACATTAC CATCGGGACA CCCCCACAGG AGTTTCAGGT GGTCTTCGAT ACTGCCTCAA GTGATCTCTG GGTGCCCAGC GATTTCTGTA CCAGCCCCGC TTGCAGCACC CATGTGCGGT TTAGACACCT GCAGAGCAGC ACCTTTAGGC TGACAAATAA GACTTTCCGG ATCACCTATG GGTCCGGAAG AATGAAAGGG GTCGTGGTGC ACGACACCGT GCGGATCGGA AACCTGGTGA GTACTGATCA GCCCTTCGGA CTGAGCATCG AGGAATACGG GTTTGAGGGA CGCATCTACG ATGGGGTGCT GGGATTGAAC TACCCCAACA TTTCCTTTTC CGGGGCAATT CCTATCTTCG ATAAGCTCAA AAACCAGAGG GCCATTTCCG AACCTGTCTT CGCCTTTTAC CTGAGCAAGG ACGAGAGGGA GGGCTCCGTC GTGATGTTTG GCGGCGTCGA TCATCGGTAC TACGAGGGCG AACTCAACTG GGTCCCCCTC ATCCAGGCCG GGGACTGGTC CGTGCACATG GACCGGATCT CCATTGAGCG GAAGATTATC GCCTGCTCCG ACGGCTGCAA GGCTCTCGTG GACACTGGAA CTAGCGATAT TGTCGGGCCT AGGCGGTTGG TGAACAACAT TCACCGGCTC ATCGGGGCCA TTCCAAGGGG CTCCGAGCAC TACGTCCCAT GCAGCGAGGT CAACACCTTG CCAAGCATTG TCTTCACAAT CAACGGCATC AACTACCCTG TCCCTGGCCG GGCATACATT TTGAAGGACG ATCGGGGCAG GTGCTACACA ACATTCCAGG AGAACAGGGT GAGCTCATCT ACCGAGACAT GGTACCTGGG CGACGTGTTC CTGAGACTCT ACTTTTCCGT GTTCGACAGG GGCAACGACA GGATCGGCTT GGCCAGGGCT GTGCTGGAGCATCACCATCACCATCACCATCACGACTACAAAGATGACGACGATAAATAGAAGCTT。下划线分别分为EcoRI 和HindIII酶切位点,5’端斜体为Kozak序列,3’端斜体为His-Flag标签位点,粗体为连接目的蛋白和His标签的Linker序列。图1电泳结果表明,PCR成功扩增出带有酶切位点的目的基因bPAG1,片段大小约1218 bp,与预期值一致。
2.2 重组质粒PCDNA3.1-bPAG1的构建及鉴定
重组载体转化后,随机挑选8个单菌落进行PCR 鉴定,电泳结果(图2)显示,泳道 1~8 在1218 bp 左右均出现与预期大小相同的目的条带,表明重组质粒成功转化大肠杆菌感受态 TOP10。重组质粒双酶切后,在 5400和1218 bp处各有1条清晰条带(图3),分别为PCDNA3.1 (-)载体和bPAG1基因,与预期结果相符,表明重组载体构建成功。将测序结果与优化后的bPAG1基因对比分析,碱基序列完全一致,符合率为100 %,其编码的氨基酸与Genbank (protein ID:AAB53145)公布的完全一致,表明bPAG1插入片段完全正确,未发生任何碱基突变。
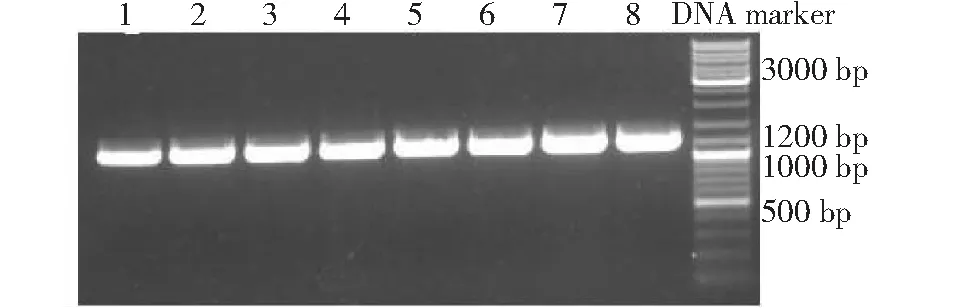
图2 阳性克隆PCR鉴定结果Fig.2 Identification of cloning products amplified by PCR
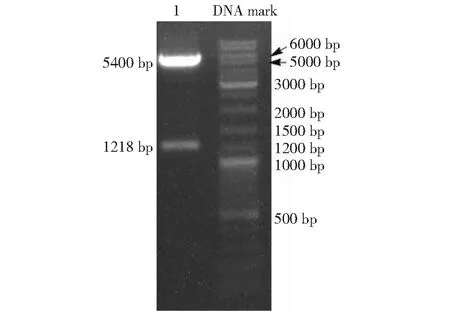
图3 重组质粒双酶切鉴定结果Fig.3 Identification of recombinant plasmid by enzymatic digestion
2.3 重组蛋白的表达纯化
重组表达载体上带有6× His标签,将诱导表达的CHO细胞上清经Ni-IDA 蛋白纯化试剂盒进行亲和层析,随后利用SDS-PAGE对纯化的蛋白进行检测,结果显示(图4),250 mM的咪唑洗脱溶液可获得纯度较高的bPAG1重组蛋白,相对分子质量约62 kDa。而bPAG1基因CDS长度为1143 bp,编码380个氨基酸残基(amino acid, AA),信号肽别区域位于第1~15位氨基酸残基,其分子式为C1925H3002N532O550S14,预测蛋白质分子量为 42.8 kDa,等电点8.98。根据实验结果可知,实际所得rbPAG1分子质量大于预期值,推测rbPAG1在表达过程中存在糖基化修饰,因为有研究表明bPAG1的氨基酸序列存在4个N-糖基化位点,导致其在翻译修饰后分子量几乎增加1倍[24]。Gel-Pro Analyzer灰度分析结果表明,纯化后的rbPAG1纯度达88 %。经BCA方法检测,纯化rbPAG1浓度为0.3 mg/mL。

M: 蛋白质分子质量标准; 1: BSA; 2: 上样液; 3: 流出液; 4: 10 mM咪唑洗脱液; 5~7: 40 mM咪唑洗脱液; 8: 80 mM咪唑洗脱液; 9~10: 250mM咪唑洗脱液; 11~13: 500mM咪唑洗脱液 M: Protein marker; 1: BSA; 2: Loading; 3: Effluent; 4: Eluent of 10 mM imidazole; 5-7: Eluent of 40 mM imidazole; 8: Eluent of 80 mM imidazole; 9-10: Eluent of 250 mM imidazole; 11-13: Eluent of 500 mM imidazole图4 SDS-PAGE分析rbPAG1纯化效果Fig.4 SDS-PAGE analysis of rbPAG1
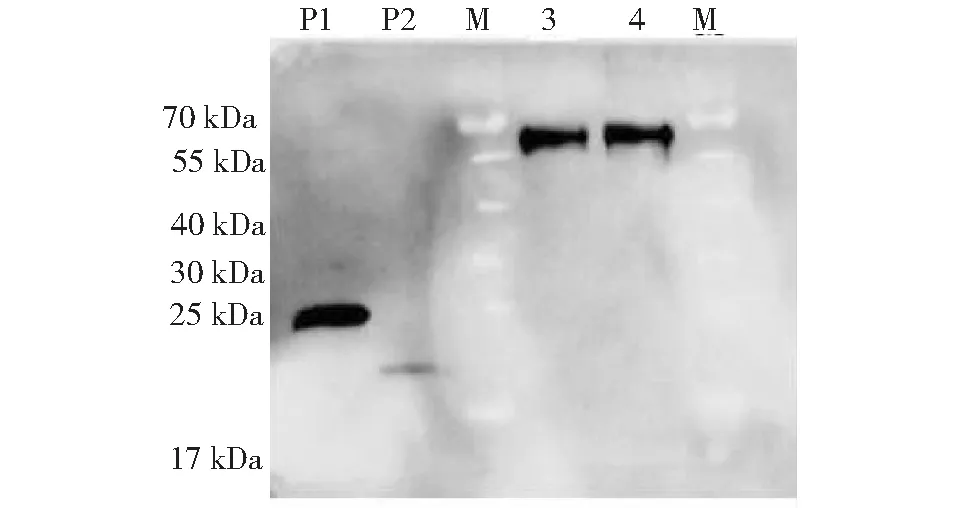
P1、P2:带His标签的阳性样本; 3, 4: 纯化后的rbPAG1(蛋白浓度0.3 mg/mL); M: 蛋白质分子质量标准P1, P2: Positive controls; 3, 4: Purified rbPAG1 (0.3mg/mL); M: Protein marker图5 rbPAG1的 Western blot鉴定Fig.5 Western blotting analysis of rbPAG1
2.4 重组蛋白的Western blot鉴定
纯化的重组蛋白C 端包含一个His 标签,根据这一特性,利用 His标签抗体对rbPAG1重组蛋白进行Western blot检测(图5)。约在62 kDa 处有特异性杂交条带,与SDS-PAGE结果一致,说明rbPAG1在真核表达系统中诱导表达成功。
3 讨 论
牛妊娠相关糖蛋白 (bPAG) 种类繁多,人们利用RT-PCR技术从牛胎盘组织中筛选出了至少22种 PAG cDNA转录本, “古代组”PAG出现在8000万年前,经过进化在约5000万年前出现“现代组”PAG[4]。 大多数 bPAG如bPAG-1、3、4、5、6、7、9、14、15、16、17、18、19、20、21、22属于“现代组”,由于催化中心碱基发生了突变而不具有酶活性;而bPAG-2、8、10、11、12、13基因属于“古代组”,在整个滋养外胚层细胞中产生,保留了典型天冬氨酸肽酶的所有特征,具有酶活性[3, 5-6]。由于bPAG家庭成员间的序列差异,bPAG基因在整个妊娠期的表达和分泌呈现时空特异性[3, 5-6, 25],如Green运用核糖核酸酶保护实验检测胎盘中RNA的表达,发现bPAG-2、4、5、8、9、10、11 早在妊娠后 25 d 就已经表达,而bPAG-1、6、7在妊娠后 45 d开始表达,主要存在于妊娠中后期[3]。
由于bPAGs 蛋白种类繁多,增加了对其结构和功能研究的难度,体外重组蛋白的出现为探究一类蛋白的结构和功能提供了新思路。因此,前人对 bPAGs 在真核系统中的表达做了相关研究,Patel 等[22](2004)构建真核表达载体 PAG-pRcRSV,分别在HEK 293和CHO细胞中首次表达了PAG1重组蛋白,但表达量较低。2010年,Telugu 等[26]为验证“古代组”bPAG基因是否具有酶活性,利用昆虫细胞杆状病毒表达系统中成功表达了bPAG2 和 bPAG12重组蛋白,并对这两种蛋白的蛋白水解活性进行研究,结果显示两个重组蛋白具有良好的蛋白水解活性。本研究为提高bPAG1在真核系统中的表达量,采用基因优化方法在不改变氨基酸序列的前提下,通过消除稀有密码子并利用偏好密码子以及平衡GC含量等策略对bPAG1基因进行设计和优化,然后通过全基因合成法合成了bPAG1优化基因,并将其成功插入到真核表达载体PCDNA3.1(-)中,采用CHO细胞经瞬时表达得到表达量及纯度均较高的bPAG1重组蛋白。本研究为后续bPAG1抗体的制备和牛早期妊娠诊断产品的研发提供了依据。
4 结 论
本研究通过全基因合成法和双酶切法将bPAG1插入到PCDNA3.1(-)表达载体中,并将重组质粒PCDNA3.1(-)-bPAG1转入CHO细胞,经过Ni柱亲和层析获得分子质量约为62 kDa的bPAG1重组蛋白,为bPAG1单克隆抗体的制备和快速检测方法的研发奠定了基础。
