成人孤立性ACTH缺乏症1例及文献复习
2019-09-25张杰于会文
张杰 于会文
成人孤立性ACTH缺乏症(adult isolated ACTH deficiency,AIAD)是一种十分少见的疾病,临床报道较少,其诊断特点是继发性肾上腺皮质功能不全,无或低皮质醇产生,除了有ACTH的低下,垂体其他激素分泌正常,垂体MRI正常或呈空泡蝶鞍,并排除有外源性糖皮质激素的使用以及垂体瘤手术后所致的ACTH缺乏[1]。常见于中、老年人,是单纯性ACTH缺乏的其中一种,其临床表现不典型,故其漏诊及误诊率较高,我院近日收治AIAD合并甲状腺功能减退症1例,现报告如下。
1临床资料
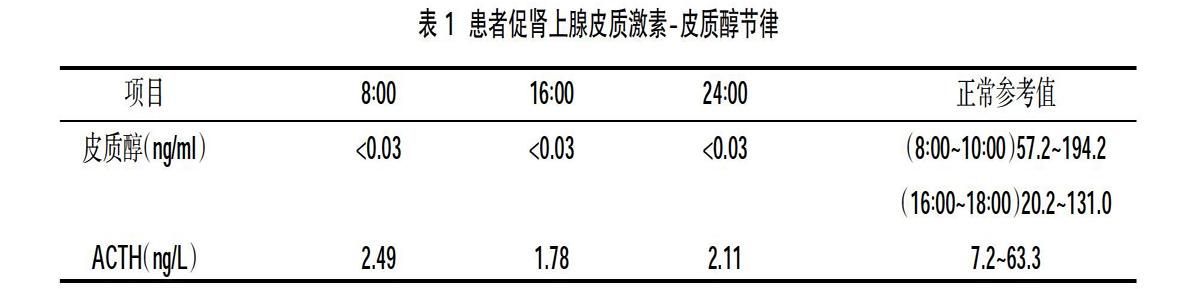
患者,男,42岁,因“恶心、呕吐3天,神志恍惚、全身冷汗1小时”于2018年4月5日入院。3 d前无明显诱因出现纳差、恶心、呕吐,腹泻,无发热,无腹痛,在外输液治疗,腹泻、呕吐症状好转,但仍感恶心、纳差。1 h前被家人发现神志恍惚、言语欠清晰,全身冷汗、在外未诊治,来我院就诊,急查随机血糖3.1 mmol/L,遂以“低血糖症”收入我科。入院查体:T 36.3℃,P 80 次/min,R 20次/min,BP 110/70 mmHg,BMI:19.38 kg/m2,体型正常,面色及全身皮肤苍白,无色素沉着,腋毛、阴毛无脱落,眼睑无浮肿,双眼视力正常,视野正常。甲状腺未扪及肿大,心肺腹无异常,双下肢无浮肿,四肢肌力、肌张力正常,双侧巴氏征阴性。无糖皮质激素应用史。辅助检查:生化全项:血糖3.11 mmol/L,钾3.9 mmol/L,钠111 mmol/L,氯80 mmol/L,总蛋白60.5 g/L,白蛋白38.2 g/L,尿素1.54 mmol/L,肌酐47.5 μmol/L,尿酸93.3 μmol/L。血常规:白细胞5×109/L,红细胞4.22×1012/L,血红蛋白119 g/L,淋巴细胞26%,嗜酸性粒细胞13.4%。甲功:TSH 49.968 μIU/ml(正常范围0.34~5.6 μIU/ml),游离T3 2.87 pmol/L(正常范围3.8~6.0 pmol/L),游离T4 5.29 pmol/L(正常范围7.4~21 pmol/L),甲状腺过氧化物酶抗体、甲状腺球蛋白抗体正常。性激素:泌乳素63.32 ng/ml(正常范围2.64~13.32 ng/ml),黄体生成素 2.63 IU/L(正常范围 1.24~8.62 IU/L),卵泡生成素 1.68 IU/L(正常范围 1.27~19.26 IU/L),雌二醇:42 pg/ml(正常范围<20~47 pg/ml),睾酮:17.24 nmol/L(6.07~27.1 nmol/L),孕1.62 nmol/L(正常范围0.32~2.67 nmol/L)。空腹胰岛素<0.3 μIU/ml。生长激素:1.5 μg/L(正常范围≤2.47 μg/ L)。甲状腺彩超:甲状腺弥漫性回声不均质。垂体磁共振未见明显异常。肾上腺CT:肾上腺萎缩表现。胸部CT未见明显异常,垂体激素测定见表1。入院诊断:成人孤立性ACTH缺乏症,给予静脉输注5%葡萄糖500 ml,给予不同剂量地塞米松静脉输注,第1~3天剂量分别为5、2.5、2.5 mg/d,第4天改为泼尼松口服,8∶00 10 mg,16∶00 5 mg,第3天加用左甲状腺素钠片50 μg/d补充甲状腺素,病情平稳,血糖、电解质恢复正常。出院后泼尼松减量为8∶00 5 mg,16∶00 2.5 mg,左甲状腺素钠片加量至100 μg/d,长期维持治疗,出院2个月后随访此患者甲状腺功能恢复正常,电解质、血糖正常。
2讨论
孤立性ACTH缺乏症的确切发病率不详,日本报道有300例以上,推算可能的患病率为 7.3/10万和 3.8/10万[2]。我国对该疾病的报道目前仍较少,可能由于该疾病的隐匿性及临床上对该病的认识不足所致。该病的临床特征有助于提高此疾病的诊断率,减少对此疾病的误诊和漏诊率。AIAD作为孤立性ACTH缺乏症中的其中一种,其病因尚不明确,主要见于成人发病。AIAD属于继发性的肾上腺皮质功能不全,目前研究结果倾向于垂体ACTH细胞选择性的损伤,从而导致了ACTH的释放和合成缺陷,可能与垂体ACTH细胞的特异性自身免疫相关[3]。
本例患者中年发病,临床特点为低钠及低氯血症、低血糖,血皮质醇及血浆ACTH 均明显降低,垂体其他激素检测水平均正常,垂体磁共振检查未见异常,符合AIAD的诊断。有报道孤立性ACTH缺乏症可合并原发性甲状腺机能减退、桥本氏甲状腺炎。该患者存在临床甲状腺机能减退,予以甲状腺素替代治疗后甲状腺功能恢复正常。
结合本文所述患者疾病特点及文献报道,总结AIAD的临床特点:①常见于中、老年人,本文报道患者42岁,生长发育正常,发病前没有糖皮质激素使用史。②首发症状不典型,多以乏力、纳差、恶心、呕吐起病,本例患者即以此症状入院。③无皮肤色素沉着,反而表现为一种皮肤色素浅淡,这一点有别于原发性肾上腺皮质功能减退症的全身色素沉着。④血浆ACTH、皮质醇水平均明显降低,但垂体其他激素未表现降低的情况。⑤血常规检查常见淋巴细胞和嗜酸性粒细胞升高,本例患者嗜酸性粒细胞升高。⑥血生化检查常有低钠低氯、轻度低血糖、血钾正常,本例患者符合此生化检查。⑦AIAD可合并多种自身免疫性疾病,且在给予糖皮质激素替代治疗后得以缓解,最常见的为自身免疫性甲状腺炎,包括桥本甲状腺炎、原发性甲状腺功能减退、Graves病等。此患者存在临床甲状腺功能减退症,予以甲状腺素替代治疗后甲功正常。⑧垂体MRI未见异常或呈空泡蝶鞍。
AIAD的治疗与继发性肾上腺皮质功能减退症的糖皮质激素替代治疗原则一致。但在应激情况下,应加大糖皮质激素剂量。当合并原发性甲状腺功能减退时,甲状腺激素的补充需要迟于糖皮质激素的替代治疗时间。因为甲状腺激素会加速皮质醇的分解,从而加重了患者的症状[4]。
总之,AIAD临床表现不典型,漏诊和误诊率很高,熟悉该病的临床特征有助于提高诊断率,减少误诊和漏诊。
参考文献:
[1]邢健,郭志新.成人孤立性ACTH缺乏症2例[J].山西医科大学学报,2016,47(12):1134-1136.
[2]Yamamoto T,Kamoi K.Prevalence of maturilv-onset isolated ACTH deficiency (IAD) in 2005:Japanese cohort studies[J].Endocr J,2008,55(5):939-941.
[3]孫婵,郑丽丽.孤立性ACTH缺乏症发病机制概述[J].河南医学研究,2015,(3):74-76.
[4]Miyauchi S,Yamashita Y,Matsuura B,et al.Isolated ACTH deficiency with Graves disease:a case report[J].Endocr J,2004,51(1):115-119.
收稿日期:2019-3-7;修回日期:2019-3-18
编辑/冯清亮
