用HPLC法测定百蕊草中3个黄酮苷的含量
2019-09-25宣伟东叶曙娜徐德东
宣伟东,成 熙,叶曙娜,徐德东
(海军军医大学附属长海医院药学部,上海 200081)
百蕊草ThesiumchinensisTurcz,为檀香科百蕊草属多年生寄生草本,别名细须草、一棵松、青龙草等,常附着在其他活体植物根上生长,在我国分布较广,主要产地为安徽、浙江和湖北等省。百蕊草性寒,味辛、涩,归肺、肾二经,有清热解毒、补肾固精之功效,现代药理研究表明,百蕊草具有良好的抗炎、解热、镇痛、抗菌等作用[1-2]。本课题组研究发现,百蕊草水提取物对肾病综合征大鼠和IgA肾病大鼠均有良好的治疗效果[3-4],其水提物经AB-8大孔树脂吸附后,中等极性的洗出物是其抗炎活性部位[5]。进一步的化学成分分离纯化研究表明,以山奈酚为母核的黄酮苷类化合物是活性部位的主要化学成分[6]。其中,含量较高的3个黄酮苷类化合物分别是:山奈酚-3-O-L-吡喃鼠李糖基(1→2)-β-D-吡喃葡萄糖苷(百蕊草素Ⅰ)、山奈酚-3-O-β-D-吡喃葡萄糖苷(紫云英苷)、山奈酚-3-O-L-吡喃鼠李糖基(1→2)-[6-O-乙酰基]-β-D-吡喃葡萄糖苷(6′-乙酰氧基百蕊草素Ⅰ)。笔者采用HPLC法,以外标法同时测定百蕊草全草中上述3个黄酮苷类化合物的含量,为百蕊草及其制剂的研究提供一种简便、快速的质量控制方法。
1 仪器与试药
LC-10AT色谱泵、SPD-10A紫外检测仪(日本岛津公司),N2000色谱工作站(浙江大学智能信息工程研究所),AB104-N电子分析天平(梅特勒-托利多)。DIKMA C18色谱柱(250 mm×4.6 mm,5 μm)。对照品百蕊草素Ⅰ、紫云英苷、6′-乙酰氧基百蕊草素Ⅰ均由本课题组从百蕊草水提取物中等极性部位分离纯化得到,HPLC纯度测定含量>98.5%[6]。3批百蕊草产地分别为:湖北荆州(安徽亳州中药材市场,批号:20150510)、安徽池州(安徽亳州中药材市场,批号:20150518)、河南泌阳(金满农场,批号:20150915),均由海军军医大学药学院中药鉴定学教研室孙莲娜副教授鉴定为檀香科百蕊草属百蕊草ThesiumchinensisTurcz的干燥全草。甲醇、乙腈为色谱纯,其余试剂均为分析纯,水为注射用水。
2 方法与结果
2.1 对照品溶液的制备
百蕊草素Ⅰ、紫云英苷、6′-乙酰氧基百蕊草素Ⅰ对照品,经105 ℃减压干燥至恒重。分别精密称定上述对照品各20、4、8 mg,分别置于100 ml量瓶中,用流动相溶解并稀释至刻度,摇匀,即得。精密量取上述3种对照品溶液各10 ml,置于同一容量瓶中,混匀,即得对照品混合溶液。
2.2 供试品溶液的制备
取干燥百蕊草全草药材,剪成1~2 cm小段。
取上述药材10 g,放入圆底烧瓶中,加入50%乙醇适量,水浴加热回流提取2次,每次1 h。合并提取液,减压回收至无醇味。浓缩液转移至100 ml分液漏斗,用20 ml水混悬,用石油醚(10 ml)萃取2次。水层减压回收成浸膏,加50%乙醇适量溶解,转移至100 ml量瓶中,再用50%乙醇稀释至刻度,摇匀。取上述溶液适量,过滤,取续滤液,即得供试品溶液。
2.3 色谱条件与系统适应性试验
色谱柱:DIKMA C18(250 mm×4.6 mm,5 μm);流动相:乙腈-水(21∶79,V/V),用冰醋酸调节pH为4.61;流速:1.0 ml/min;柱温:25 ℃;检测波长:346 nm。对照品混合溶液和供试品溶液各进样20 μl,分别记录色谱图,各组分理论塔板数均>3 000,分离度>1.5,色谱图见图1。
2.4 线性关系考察

以浓度(X,μg/ml)为横坐标,面积(Y)为纵坐标进行线性回归,得回归方程,结果见表2。

图 1 对照品及供试品溶液的HPLC图 A.混合对照品溶液;B.供试品溶液 1.百蕊草素Ⅰ;2.紫云英苷;3.6′-乙酰氧基百蕊草素Ⅰ
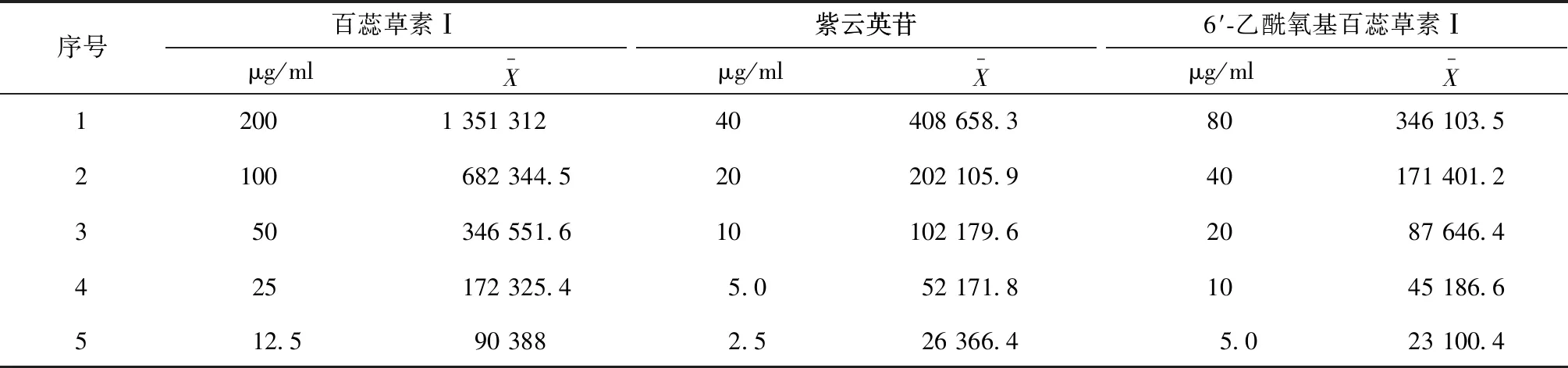
序号百蕊草素Ⅰμg/mlX-紫云英苷紫云英苷μg/mlX-6′-乙酰氧基百蕊草素Ⅰμg/mlX-1200135131240408658.380346103.52100682344.520202105.940171401.2350346551.610102179.62087646.4425172325.45.052171.81045186.6512.5903882.526366.45.023100.4

表2 各对照品线性关系考察结果
2.5 精密度试验
取线性关系考察项中序号3对照品混合溶液(百蕊草素Ⅰ:50 μg/ml、紫云英苷:10 μg/ml、6′-乙酰氧基百蕊草素Ⅰ:20 μg/ml),进样20 μl,连续进样6次,记录峰面积。对照品百蕊草素Ⅰ、紫云英苷、6′-乙酰氧基百蕊草素Ⅰ的平均峰面积分别为366 795.9、971 50.82、746 25.32,RSD(n=6)分别为1.08%、1.20%、1.77%,表明仪器精密度良好。
2.6 稳定性试验
取同一份供试品溶液Ⅰ(批号:20150510),分别于0、2、4、8、12、24 h按上述色谱条件进样20 μl,记录色谱图和各成分峰面积。供试品中百蕊草素Ⅰ、紫云英苷、6′-乙酰氧基百蕊草素Ⅰ的平均峰面积分别为408 302.5、107 108.6、904 45.45,RSD(n=6)分别为0.85%、1.32%、2.09%,表明供试品溶液在24 h内稳定性良好。
2.7 重复性试验
取同一批次百蕊草药材6份(批号:20150510),每份各约10.0 g,按“2.2”项下方法制备供试品溶液,各进样20 μl,分别记录色谱图和各成分峰面积。各供试品中百蕊草素Ⅰ、 紫云英苷、 6′-乙酰氧基百蕊草素Ⅰ的平均峰面积分别为335 104.2、110 128、819 38.33,RSD(n=6)分别为2.43%、2.22%、1.72%,表明本方法重复性良好。
2.8 加样回收率试验
依据重复性考察结果,按外标法计算10.0 g百蕊草中(批号:20150510)百蕊草素Ⅰ、紫云英苷、6′-乙酰氧基百蕊草素Ⅰ的含量分别为:45.7、1.1、2.2 mg。取上述百蕊草药材6份,每份约5.0 g,分别置于圆底烧瓶中,每份中均加入干燥的对照品百蕊草素Ⅰ(22.9 mg)、紫云英苷(0.55 mg)和6′-乙酰氧基百蕊草素Ⅰ(1.1 mg),按“2.2”项下方法平行制备供试品溶液并按上述色谱条件进样20 μl进行测定,记录峰面积并计算各成分平均回收率(n= 6)。结果见表3。
2.9 样品含量测定
取3批百蕊草药材(批号:20150510、20150518、20150925),按“2.2”项下方法制备供试品溶液。供试品溶液和对照品混合溶液各进样20 μl测定,记录峰面积。采用外标法分别计算3批百蕊草药材中百蕊草素Ⅰ、紫云英苷、6′-乙酰氧基百蕊草素Ⅰ的含量。结果见表4。

表3 加样回收率试验测定结果(n=6)

表4 百蕊草中3种成分含量测定结果(n=3)
3 讨论
文献报道的百蕊草药材以及上市制剂“百瑞含片”的质量标准中[7-8],多以山奈酚为测定指标。因山奈酚在百蕊草中含量极低,故采用酸水解法提取药材后制备供试品溶液,以提高山奈酚的含量。经酸水解之后制备的供试液中,山奈酚的含量已非药材天然含有的水平,不能真实反映药材及制剂中真实的含量。且酸水解为化学反应,可带来不可预知的副反应及其他产物,水解时的pH值、时间和温度等因素均会导致水解程度不同,影响含量的准确性。有报道提示,选用50%乙醇溶液回流提取2次,每次1 h,紫云英苷的提取率最高[7]。本实验测定的3个黄酮苷类化合物为紫云英苷及其衍生物,结构中均含有糖分子,在含水溶剂中溶解度较大,不溶于低极性的有机溶剂。因此,在供试品溶液制备时,选用50%乙醇溶液提取2次,并用石油醚萃取提取液,去除极性较小的成分,从而排除脂溶性成分对液相色谱的干扰。
通过对混合对照品溶液的紫外扫描,分别在265、346、378 nm处出现山奈酚的特征吸收峰。由于265 nm处多数成分均有吸收,专属性不强,对基线干扰大,而368 nm的吸光度较小,故采用346 nm作为检测波长,可以获得分离度较好的HPLC图谱。基于有文献采用一测多评法测定百蕊草中山奈酚、百蕊草素Ⅰ及百蕊草素Ⅱ的含量,采用相对校正因子计算和外标法测定3个成分的含量无显著差异[9],故本实验采用外标法直接测定含量。
在百蕊草活性成分分离纯化的基础上,以3个黄酮苷化合物为对照,采用HPLC法,对湖北、安徽、河南产地的3批全草进行了活性成分的含量测定,首次建立了一次同时测定百蕊草中3个黄酮苷类组分的方法,可为百蕊草及其制剂的质量控制提供一种新的、简便、快速的分析手段。
