紫花苜蓿耐苏打盐碱相关基因的转录组学分析
2019-09-25邬婷婷方志坚焦德志孙婴宁
李 红, 李 波, 邬婷婷, 杨 曌, 方志坚, 焦德志, 孙婴宁
(1. 黑龙江省农业科学院畜牧兽医分院, 黑龙江 齐齐哈尔161005; 2.齐齐哈尔大学生命科学与农林学院,抗性基因工程与寒地生物多样性保护黑龙江省重点实验室, 黑龙江 齐齐哈尔 161006)
紫花苜蓿(MedicagosativaL.)是世界上栽培最多的多年生豆科植物之一,因其具有高产,营养价值高,适应性广等特点,故称“牧草之王”[1-2]。生长中的植物常受盐碱等非生物胁迫,因此,植物发展了各种复杂的机制来适应这种环境[3-4]。逆境胁迫时,植物在生理生化和分子水平上均发生改变,植物生理响应包括了光合能力、光合途径、发育阶段的调整、渗透调节、抗氧化防御及积累相关胁迫功能蛋白等代谢进程的变化。这些生理性状均随植物自身发育进程与环境不同而改变。植物在次生代谢物水平上通过蛋白与蛋白、蛋白与核酸、蛋白与脂类相互作用及累积渗透调节物质来提高抗逆性[5]。
近年来,对紫花苜蓿耐盐碱基因的研究主要集中在基因克隆和转录组分析两方面,例如:Luo等[6]利用RT-PCR技术从盐处理下的紫花苜蓿中分离出解旋酶基因MH1,其中甘露醇、NaCl、甲基紫精和脱落酸能够诱导MH1表达。通过对转基因拟南芥(Arabidopsisthaliana)中渗透调节物质和过氧化物酶的检测证明该基因能够提高植物渗透调节和活性氧(Reactive oxygen species,ROS)的清除能力。Postnikova等[7]对盐胁迫下紫花苜蓿的根进行转录组分析,耐盐型和盐敏感型品种通过GO注释得到367 619 586条reads。生物信息学分析表明,盐胁迫下共1 165 个基因(含86个转录因子)发生了改变。与拟南芥的GO注释比对后有40%的基因差异表达。盐碱处理以中性盐、单一碱性盐为主,而对复合苏打盐碱胁迫下紫花苜蓿抗逆基因缺乏系统性论述,土壤中NaHCO3和Na2CO3含量过高会对植株造成离子毒害作用,紫花苜蓿作为改良盐碱地土壤的重要牧草,在恢复盐碱地植被种植、降低土壤pH、提高盐碱地牧草产量和农业化生产等方面具有重要意义[8]。本研究利用RNA测序(RNA sequencing,RNA-Seq)对混合苏打盐碱胁迫前后的紫花苜蓿叶片进行转录组测序,筛选出紫花苜蓿叶片响应混合苏打盐碱胁迫下的差异表达基因(Differential-expressed genes,DEGs)。对差异表达基因进行NCBI非冗余蛋白库(Non-redundant,NR)、基因本体(Gene Ontology,GO)数据库、蛋白直系同源簇数据库(Clusters of Orthologous Groups of proteins,COG)和京都基因和基因组百科全书公共数据(Kyoto Encyclopedia of Genes and Genomes,KEGG)注释和富集分析,从转录组水平研究紫花苜蓿耐苏打盐碱机制,为抗盐紫花苜蓿的研究提供依据。
1 材料与方法
1.1 试验材料及处理
以紫花苜蓿WL343HQ幼苗为试验材料,首先对紫花苜蓿WL343HQ种子进行消毒处理10 min,然后用灭菌水清洗3次,在灭菌水中浸泡2 h,播种到育苗盆中。待幼苗长至三叶一心期时,将生长整齐基本一致的紫花苜蓿幼苗取出用清水洗净根部,加入1/2改良霍格兰营养液预培养(3 d)。将紫花苜蓿幼苗分为2组,其中WL-CK组用无菌蒸馏水培养72 h,WL-EG组用150 mmol·L-1的NaHCO3,Na2CO3混合溶液(NaHCO3∶Na2CO3=9∶1,pH=9.11)胁迫(72 h)。取长势一致叶片大小相近的2组紫花苜蓿叶片于冰箱中冷冻保存(—80℃),用于转录组测序,生物学重复3次。
1.2 总RNA提取、检测及Illumina测序
采用TaKaRa公司的MiniBEST plant RNA Extration Kit提取紫花苜蓿叶片总RNA。经琼脂糖凝胶电泳和Agilent 2100对实验组、对照组的总RNA浓度进行检测。由紫花苜蓿叶片提取出的总RNA通过Illumina公司的TruseqTM RNA sample prep Kit获得mRNA,并反转录成cDNA,经过2%琼脂糖凝胶回收、TBS380定量及cBot桥式PCR扩增形成clusters,在Hiseq4000测序平台进行双端测序。Base Calling测序得原始图像,先转化为Raw Data,去除低质量读段后转化为Clean Data,使用Trinity(http://trinityrnaseq. sourceforge. net/ analysis/ extract proteins from trinity transcripts.html)软件[16]对所有Clean Data进行从头组装。
1.3 差异基因分析方法
紫花苜蓿叶片转录组数据库组装完成后,采用BWT(Burrows-Wheeler Transform) 算法的Bowtie(http://bowtie-bio.Sourceforge.net/index.shtml)[9]将测序得到的clean reads对应到组装后得到的转录组上。通过RSEM(RNA-Seq by Expectation Maximization,http://deweylab.biostat.wisc.edu/rsem/)[10]软件来精确衡量Denovo组装的完整性,利用每千碱基外显子百万片段数FPKM (Fragments per kilobase million)[11]来对PE测序上的RPKM(Reads per kilobase million)[12]进行校正并计算基因差异表达倍数。利用edge R(Empirical analysis of Digital Gene Expression data in R,http://www.bioconductor.org/packages/release/bioc/html/edge R.html)[13]识别此次转录本数据差异表达的基因。用多重校验方法对差异表达基因的阈值p-value进行校正后得错误发现率(False discovery rate,FDR)值[14]。我们用FDR<0.05 和∣log2FC≥1∣作为判断和筛选显著差异表达基因的条件。使用BlastX分别与NR、COG、GO、KEGG数据库进行比对获得注释信息,使用Goatools (https://github.com/tanghaibao/GOatools)[15],对经过校正的p值(p_FDR)≤0.05 时,认为此GO功能存在显著富集情况。对于通路的富集分析,使用KOBAS(http://kobas.cbi.pku.edu.cn/home.do)[16],把满足阈值P-Value<0.05条件的通路定义为在DEGs中显著富集的KEGG通路。
1.4 SSR位点分析
对紫花苜蓿转录组中的Unigene序列进行简单重复序列(Simple sequence repeats,SSR)位点分析,采用MISA(http://pgrc.ipk-gatersleben.de/misa/misa.html)软件[17](1.0 版,默认参数:对应的各个unit size的最少重复次数分别为 1-10,2-6,3-5,4-5,5-5,6-5)对Unigene进行 SSR检测。
1.5 qRT-PCR验证
提取对照组和处理组的叶片总RNA,采用PrimeScript RT reagent Kit with gDNA Eraser试剂盒反转录成cDNA,进行qRT-PCR分析。随机选取5个差异表达基因,利用Primer Premier 3.0软件设计基因的正反向引物(见表1),以YHL EF-1α为内参基因,各处理均3次重复并计算基因表达量。
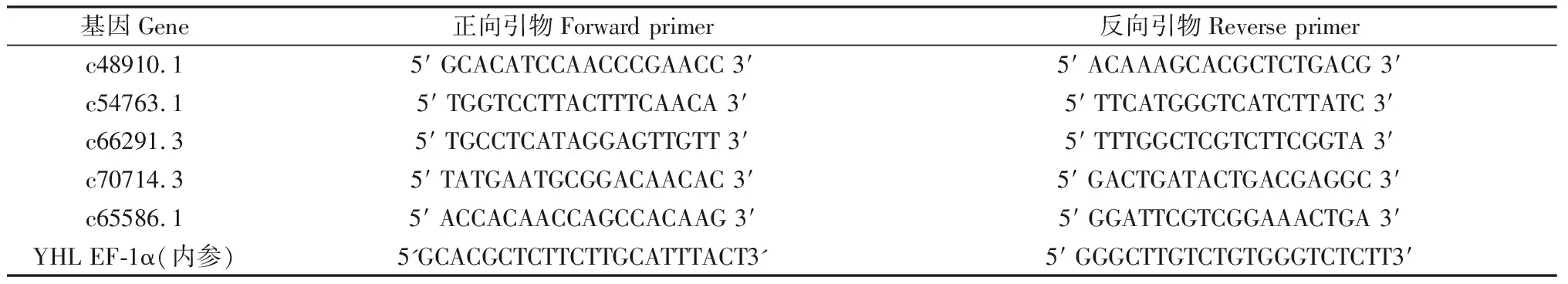
表1 DEGs引物信息Tab 1 DEGs primer information
2 结果与分析
2.1 转录组测序、De novo组装和序列分析
如表2所示,对试验中的2个样品测序后分别获得了8千万余条原始序列数据,过滤后Clean data占原始序列数据95.24%和96.44%;碱基数占原始碱基数的88.23%和92.04%;两个样品碱基错误率分别由0.0378%降低到0.0268%和0.0238%到0.0184%,GC%含量变化不大。测序分析中把Q≥20的碱基作为高质量,本研究中Q20值在 96.37%~90.15%之间。混合2个样品的高质量读段,用Trinity软件[18]进行从头组装,denovo组装后共得到91 853 个Unigenes,每个Unigenes的长度为711.67 bp。

表2 测序数据统计结果Tab 2 Output statistics of sequencing
Unigenes长度统计如图1所示。可知,这些Unigenes中,序列长度在1 ~ 1 000 bp,1 001 ~ 2 000 bp,2 001 ~ 3 200 bp,3 201~ 5 000 bp之间的序列条数分别为70 016 (81.23%),11 465 (12.49%),4 151 (4.52%),1 175 (1.28%),仅445条(0.48%)序列长度大于5 000 bp,从图中Unigenes长度分布可以看出,随着序列长度的增加,序列数逐渐减少,整体呈现下降的趋势,说明组装良好。
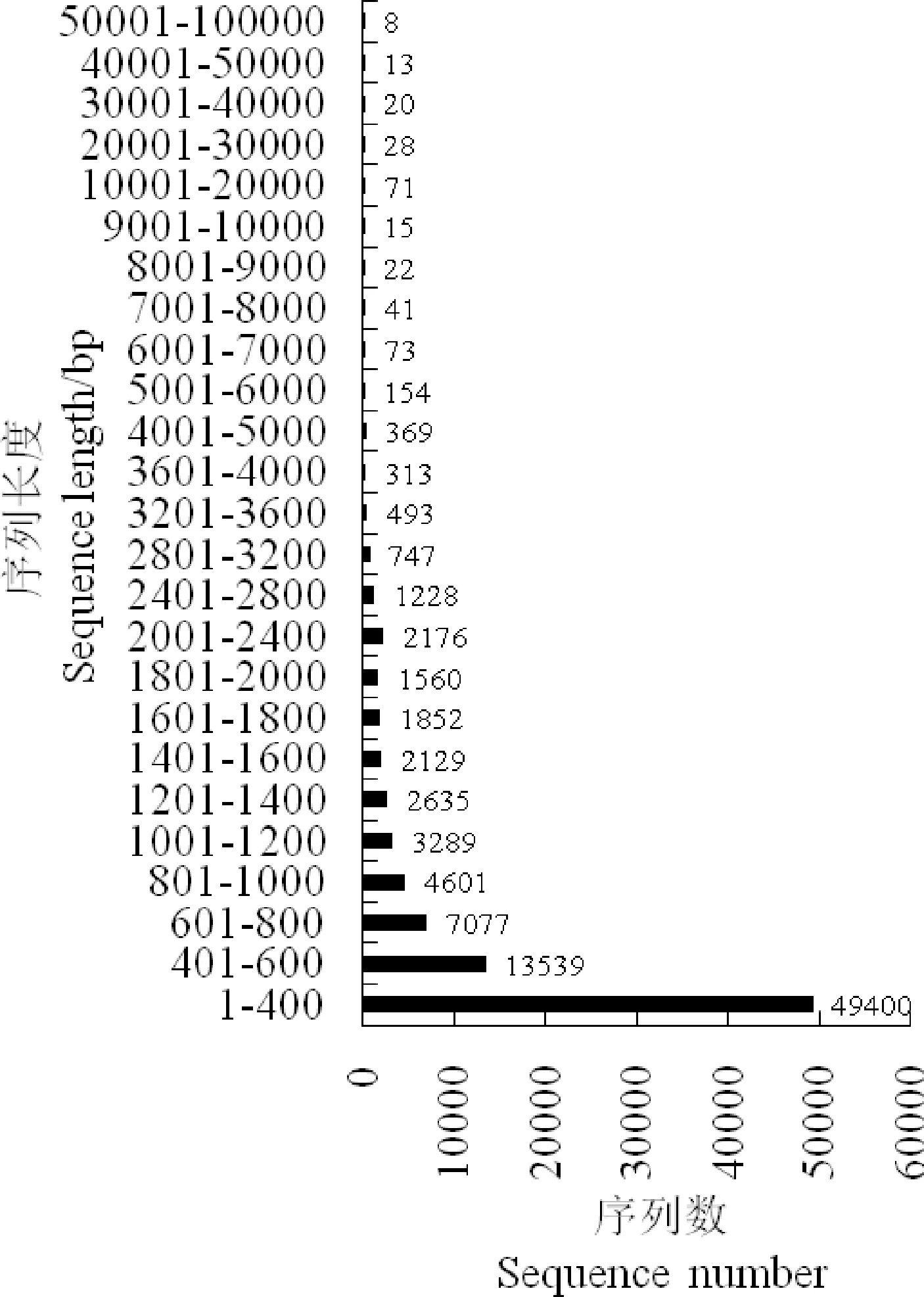
图1 组装后Unigenes长度分布统计图Fig.1 Unigenes length distribution chart after assembly
2.2 NR库注释结果分析
如图2(A)所示,45 540个Unigenes与NCBI中NR数据库比对中获得注释,其中24 765(54.381%)Unigenes与之完全匹配,少数Unigenes不完全匹配。图2(B)中,相似度在80%~100%,60%~80%和40%~60%的Unigenes数及所占比例分别为38 886(85.389%),6 128(13.456%),525(1.153%)。如图2(C),被注释到蒺藜苜蓿(Medicagotruncatula)、鹰嘴豆(Cicerarietinum)、小鼠(Musmusculus)、大豆(Glycinemax)、拟南芥(Arabidopsisthaliana)、葡萄(Vitisvinifera)、紫花苜蓿(MedicagosativaL.)、硫微螺菌属(Thiomicrospirasp. MA2-6)、菜豆(Phaseolusvulgaris)、其他物种和未知物种上的Unigenes数所占比例分别为62.716%,3.276%,1.968%,1.296%,1.170%,0.773%,0.558%,0.547%,0.505%,6.447%和20.744%,其中被注释到蒺藜苜蓿上的Unigenes所占比例最大。

图2 NR功能分类Fig. 2 Classification of NR functions注:图2(C)中Ⅰ-Ⅺ分别代表蒺藜苜蓿、鹰嘴豆、小鼠、大豆、拟南芥、葡萄、紫花苜蓿、硫微螺菌属、菜豆、其他物种和未知物种Note:Fig 2(C):I-XI represents Medicago truncatula, Cicer arietinum, Mus musculus, Glycine max,Arabidopsis thaliana,Vitis vinifera,Medicago sativa L.,Thiomicrospira sp. MA2-6,Phaseolus vulgaris,other species and unknown species
2.3 COG注释结果分析
为了进一步评估苜蓿叶片Unigenes的完整性并预测其功能,现将Unigenes与NCBI中COG数据库进行比对。结果发现,共13 141个Unigenes被注释归纳到25个COG功能族上(图3),包含Unigenes数最多的5个代谢途径分别是:仅一般功能预测(General function prediction only),信号转导机制(Signal transduction mechanisms),翻译后修饰,蛋白质折叠,分子伴侣(Posttranslational modification,protein turnover,chaperones),翻译,核糖体结构与生物发生(Translation,ribosomal structure and biogenesis),碳水化合物运输和代谢(Carbohydrate transport and metabolism),这些代谢途径中Unigenes数及所占比例分别为1 034(7.869%),1 020(7.762%),810(6.164%),722(5.494%),568(4.322%)。包含Unigenes数最少2个功能族有胞外结构和核结构,仅有1条,其余Unigenes则被注释到RNA加工与修饰(RNA processing and modification),染色体结构与活力(Chromatin structure and dynamics),防御机制(Defense mechanisms),细胞运动(Cell motility)等途径。

图3 COG功能分类Fig. 3 Classification of COG functions
2.4 GO注释结果分析
GO数据库共包括三个本体,分别描述分子功能(Molecular function)、细胞组分(Cellular compontent)及生物学过程(Biological process)。对Unigenes进行GO分类结果表明(图4),盐碱处理下的紫花苜蓿叶片,共得到5 484个差异表达基因,其中2 688个Unigenes上调,2 796个Unigenes下调,5 484个Unigenes可分为50个功能组,其中包含代谢过程(Metabolic process,up 281,down 282),细胞过程(Cellular process,up 248,down 249),单一生物过程(Single-organism process,up 205,down 200),刺激应答过程(Response to stimulus,up 101,down 91),生物调节(Biological regulation,up 80,down 74)等23个生物学过程,细胞(Cell,up 196,down 211),细胞器(Organelle,up 138,down 169),膜(Membrane,up 96,down 109),细胞器组分(Organelle part,up 77,down 88)等15个细胞组分,结合(Binding,up 253,down 240),催化活性(Catalytic activity,up 222,down 223),转运活性(Transporter activity,up 21,down 21),结构分子活性(Structural molecule activity up,13,down 15),核酸结合转录因子活性(Nucleic acid binding transcription factor activity,up 7,down 8)等12个分子功能。
2.5 KEGG注释结果分析
KEGG通路数据库中包含了在细胞内的分子相互作用的信息网络和具体的变化路径[19-20]。通过与KEGG数据库的比较,可以更好的了解在盐碱胁迫诱导下苜蓿叶片中表现较为活跃的生物通路。代谢途径(Metabolic pathways)(3 504)是被注释到最多序列的通路,其次是次生代谢产物的生物合成(Biosynthesis of secondary metabolites)(1 865)、抗生素合成(Biosynthesis of antibiotics)(823)、不同环境中微生物代谢(Microbial metabolism in diverse environments)(710)、碳代谢(Carbon metabolism)(514),除上述五大代谢途径外,包含Unigenes最多的前20个代谢通路如图5所示,包含核糖体(Ribosome),Epstein-Barr病毒侵染(Epstein-Barr virus infection),蛋白质在内质网加工(Protein processing in endoplasmic reticulum)等途径。

图4 GO功能分类Fig.4 GO function classification

图5 Unigenes最多的前20个通路Fig.5 The top 20 paths contain most unigenes
2.6 Unigenes的通路注释结果分析
将KEGG数据库作为参考,将Unigenes具体定位到26个主要代谢途径中(表3),其中包含unigenes最多的5个分别为代谢途径(Metabolic pathways,284,12.09%)、次生代谢物的生物合成(Biosynthesis of secondary metabolites,154,6.56%)、微生物在不同环境中的代谢(Microbial metabolism in diverse environments,46,1.96%)、核糖体代谢途径(Ribosome,46,1.96%)、抗生素生物合成(Biosynthesis of antibiotics,46,1.96%),而错配修复(Mismatch repair)、同源重组(Homologous recombination)、谷胱甘肽代谢(Glutathione metabolism)、核苷酸切除修复(Nucleotide excision repair)、半胱氨酸和蛋氨酸代谢(Cysteine and methionine metabolism)等途径包含DEGs数较少,为15个,占注释总基因数的0.64%。其余则被包含在光合作用途径(Photosynthesis,43)、植物激素信号转导途径(Plant hormone signal transductio,41)、碳代谢途径(Carbon metabolism,38)、苯丙素生物合成途径(Phenylpropanoid biosynthesis,36)等。
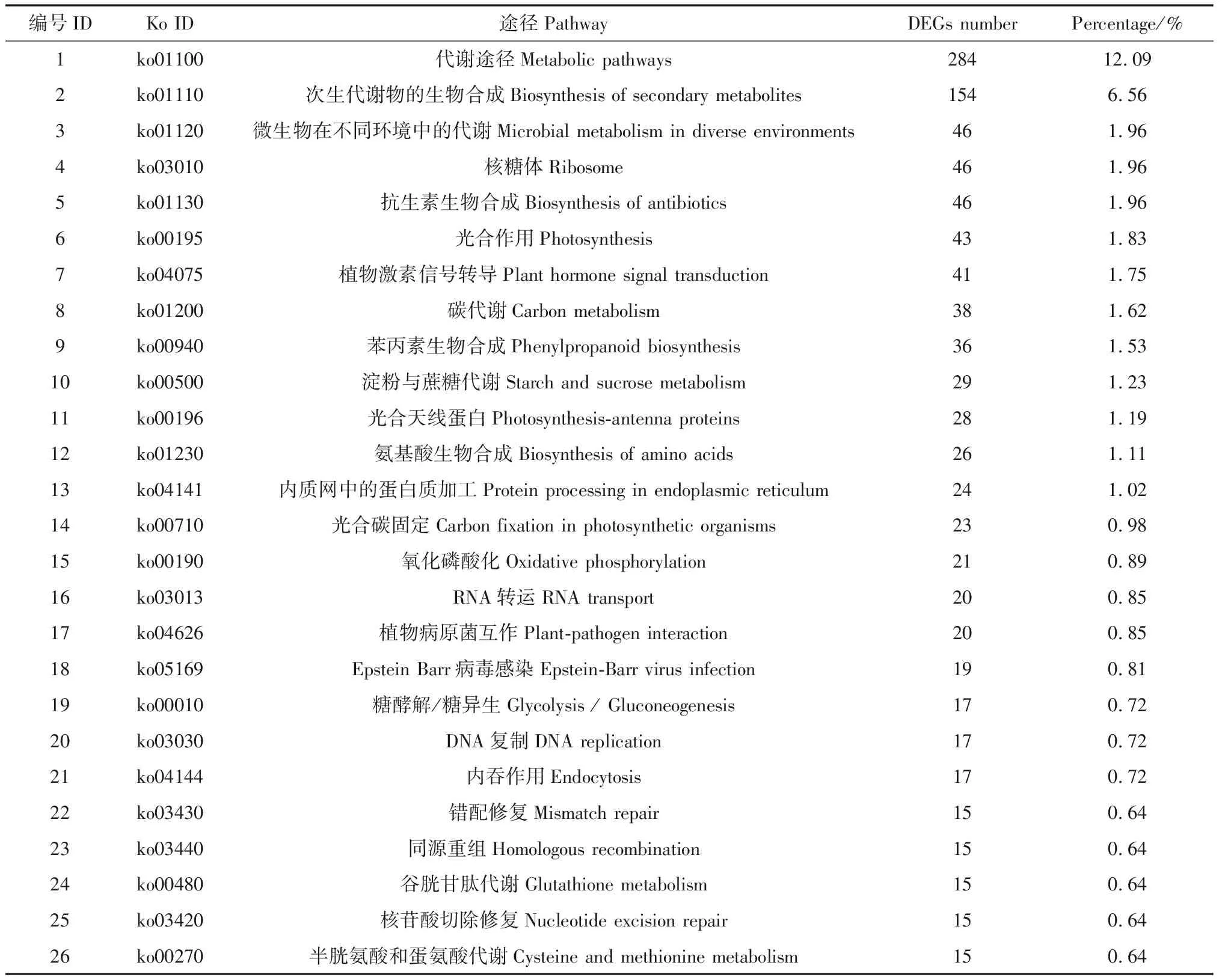
表3 Unigenes通路注释结果统计Tab 3 Statistical results of unigenes pathway annotation
2.6 紫花苜蓿苏打盐胁迫应答基因及转录因子分析
利用BLASTP对KEGG注释通路显著性分析,发现许多和苏打盐碱胁迫相关的DEGs及转录因子(如表4),如木质素生物合成相关基因(c66804.1,c65832.1,c60601.1等),激素信号转导途径相关基因(c62547.1,c60820.1,c37217.1等),谷胱甘肽代谢途径相关基因(c65962.1,c61345.1,c54763.1等),参与光合作用过程相关基因(c19710.1,c83213.1,c52958.1等),乙烯应答转录因子(c62547.1),EREBP亚家族转录因子(c74585.1,c64644.1),同源异型域转录因子(c50130.1,c66549.1),MYB转录因子(c64519.1,c52531.1,c48627.1)及HSF转录因子(c51789.1,c41124.1,c29594.1等)。

表4 紫花苜蓿响应苏打盐碱胁迫诱导表达重要基因及转录因子Tab 4 Alfalfa induced expressions of important genes and transcription factor in response to soda saline-alkali stress
2.7 SSR位点分析结果
对紫花苜蓿筛选出的91 853个Unigenes通过SSR位点分析得到10 949个SSR位点(图6),包含6种核苷酸基序类型,单核苷酸基序有5 278个,占比最高达到48.205%,五核苷酸基序和六核苷酸基序所占比例最低,分别为0.192%和0.110%。如图7,在检测到的SSR核苷酸基序中出现频率最高的10类基序及所占比例分别为A/T(5 240个,47.858%)、AC/GT(584个,5.334%)、AG/CT(1 422个,12.987%)、AT/AT(313个,2.859%)、AAC/GTT(522个,4.768%)、AAG/CTT(1 007个,9.197%)、AAT/ATT(291个,2.658%)、ACC/GGT(308个,2.813%)、AGG/CCT(161个,1.470%)、ATC/ATG(530个,4.841%),此SSR特征有助于紫花苜蓿基因组差异分析以及遗传图谱的构建。

图6 核苷酸基序分类图Fig.6 Classification of nucleotide motifs

图7 SSR中出现频率最高的10类基序 Fig.7 The most frequently occurring 10 types of motifs in SSR
2.8 差异表达基因主要转录因子家族分析
转录因子是转录调控的核心功能蛋白。紫花苜蓿转录组数据分析GH3、MYB、HSF、HD-ZIP、4CL、EREBP、WRKY、ERF等转录因子家族与苏打盐碱胁迫高度相关,其中MYB、HSF、HD-ZIP等转录因子家族包含基因数较多,EREBP、WRKY、ERF等转录因子家族包含基因数较少。

图8 紫花苜蓿叶片响应苏打盐碱胁迫差异基因转录因子Fig.8 Transcription factors differentially expressed in leaves of alfalfa under soda saline-alkali stress
2.9 差异表达基因qRT-PCR验证结果分析
为了验证差异基因数据的可靠性,随机选取5个差异表达基因,其中4个基因上调,1个基因下调,qRT-PCR荧光定量结果表明,5个基因在苏打盐碱胁迫胁迫后的表达趋势与RNA-Seq结果一致(图9),说明测定转录组数据的可靠性。

图9 差异表达基因qRT-PCR验证Fig.9 Verification of differentially expressed genes by qRT-PCR注:A、B、C、D、E分别代表c48910.1、c54763.1、c66291.3、c70714.3、c65586.1Note:A、B、C、D、E significant c48910.1、c54763.1、c66291.3、c70714.3、c65586.1
3 讨论
转录组学(transcriptomics) 是功能基因组学研究的一个重要内容,它是从整体水平上研究细胞中基因转录的情况及其转录调控规律。随着新一代测序技术的发展,其高通量性、低成本、快速、准确的特点可大大提高测序的效率,降低试验成本,为RNA水平研究基因表达提供有效方法。
在盐碱等非生物胁迫下,植物通过基因的改变来调控代谢及信号转导途径,从转录和翻译等水平上作出响应,逆境胁迫下的转录因子会激活或抑制下游基因的表达[21]。本研究中鉴定出GH3、MYB、HSF、HD-ZIP、4CL、EREBP、WRKY、ERF等转录因子与紫花苜蓿耐盐碱性相关,说明紫花苜蓿耐盐碱性应答机制是由多种转录因子共同调控。
ABA(Abscisic acid)信号转导通过PYR/PYL/RCAR蛋白与蛋白磷酸酶2C(Protein phosphatase 2C,PP2C)家族的A类蛋白结合,释放磷酸化蔗糖非酵解型蛋白激酶2(Sucrose non-fermenting 1-related protein kinase2),从而激活ABA信号转导下游基因的表达[22]。本研究中共鉴定出5个PP2C家族基因,其中4个上调,1个下调,推测该基因在紫花苜蓿逆境胁迫中起关键性作用。生长素早期应答基因分为Aux/IAAs,GH3s,SAURs三种,参与植物顶端优势、根部形成和逆境胁迫等过程[23]。GH3蛋白具有吲哚乙酸(Indole-3-acetic acid,IAA)和茉莉酸(Jasmonic acid,JA)等激素氨基酸化合成酶功能,参与植物光合作用途径[24]。本研究中,c60820.1,c37217.1,c66609.1等差异表达基因全部上调,说明GH3家族基因在植物防卫反应过程起重要作用。
AP2/EREBP转录家族包含AP2和EREBP两个亚族,与植物的逆境响应有关,其中ERF转录因子会激活抗氧化基因的表达,减轻植株氧化损伤[25-26]。本研究共鉴定出2个下调EREBP基因和2个上调ERF基因,说明ERF基因主要通过增强乙烯的生物合成来增强紫花苜蓿的耐盐性,即两类基因均参与紫花苜蓿盐碱胁迫应答过程,但调控方式不同。
WRKY转录因子家族是植物特有的转录调控因子,紫花苜蓿在应对非生物胁迫过程中,WRKY转录因子家族基因过表达以适应逆境环境[27]。例如:拟南芥中过表达WRKY25和WRKY33基因能提高耐盐性,过表达WRKY46,WRKY54,WRKY70能提高植株对干旱的耐受性[28-29]。本研究仅鉴定出1个WRKY基因,说明WRKY转录因子在响应苏打盐碱胁迫过程中作用较小。
苏打盐碱胁迫产生的活性氧会对植株造成氧化损伤,研究表明盐胁迫下拟南芥谷胱甘肽S转移酶(Glutathione S-transferase,GST)和谷胱甘肽过氧化物酶(Glutathione peroxidase,GPX)基因均上调表达[30]。本研究中共鉴定出9个谷胱甘肽s转移酶基因,7个上调表达,2个下调表达,这些基因均参与ROS的清除系统,减缓苏打盐碱胁迫下紫花苜蓿幼苗叶片的氧化胁迫,从而增强其耐盐碱性。小家族热激转录因子(HSF)对植物提高非生物胁迫的耐受性起重要作用[31]。本研究共鉴定出16个HSF转录因子家族基因,其中10个上调表达,6个下调表达,说明HSF家族转录因子与盐碱胁迫应答具一定的关联性。
4 结论
本试验采用RNA-Seq技术筛选出许多与盐碱胁迫有关的差异表达基因,其中2 688个上调基因和 2 796 个下调基因。发现GH3,MYB,HD-ZIP,EREBP,WRKY,ERF等转录因子与紫花苜蓿耐盐碱性相关。通过GO注释和KEGG注释分析发现,这些基因主要参与核糖体途径、光合作用途径、植物激素信号转导途径、氧化磷酸化途径、谷胱甘肽代谢途径、苯丙氨酸代谢途径等许多代谢途径,但我们对这些基因对代谢途径的调控机制了解甚少,需深入研究,从而进一步了解紫花苜蓿盐碱胁迫调控机制。
