3228例疑似遗传代谢病串联质谱检测结果和临床分析
2019-09-17杨潍嘉蒋庆安巫玉峰黄开明胡卫秦盛贝
杨潍嘉 蒋庆安 巫玉峰 黄开明 胡卫 秦盛贝

【摘要】 目的 探讨串联质谱技术在新生儿疾病筛查和高危儿筛查中的意义,初步了解桂林市遗传代谢病的发病率及病种分布情况。方法 对2018年4月~2019年5月桂林市妇女儿童医院出生的新生儿和门诊或者住院部疑似遗传代谢病患儿共3228例,应用串联质谱技术进行遗传代谢病筛查,对初筛阳性或者可疑病例召回复查并结合其他检测进行临床分析。结果 3228例中,可疑阳性171例,筛查阳性率5.30%,确诊遗传代谢病7例(2.17‰)。分别是苯丙氨酸羟化酶缺乏症3例(0.93‰),母源性肉碱缺乏症1例(0.31‰),同型半胱氨酸血症1例(0.31‰),糖原累积症2例(0.62‰)。结论 本研究检出的遗传代谢病发病率较高,检出病种有限,需进一步进行全员、大样本量覆盖到桂林市每个县的新生儿筛查中。
【关键词】 串联质谱技术;遗传代谢病
中图分类号:R725.8 文献标志码:A DOI:10.3969/j.issn.1003-1383.2019.08.004
【Abstract】 Objective To study the significance of tandem mass spectrometry in the screening of neonatal diseases and high-risk infants,so as to preliminarily understand the incidence and distribution of genetic metabolic diseases in Guilin.Methods From April 2018 to May 2019,3228 newborns who were born in Guilin Women and Children Hospital and children with suspected genetic metabolic diseases in outpatient or inpatient departments were enrolled.The genetic metabolic diseases were screened by tandem mass spectrometry,and positive or suspicious cases were recalled for reexamination and clinical analysis in the combination of other tests.Results Of the 3228 cases,171 were suspicious positive,the positive rate of screening was 5.30%,and 7 cases(2.17‰) were diagnosed as hereditary metabolic diseases.There were 3 cases(0.93‰) of phenylalanine hydroxylase deficiency,1 case(0.31‰) of maternal carnitine deficiency,1 case(0.31‰) of homocysteinemia,and 2 cases(0.62‰) of glycogen accumulation.Conclusion The incidence of hereditary metabolic diseases detected in this study is high,and the types of diseases detected are limited.It is suggested that more extensive publicity should be given to cover the sample size in the neonatal screening in every county of Guilin.
【Key words】 tandem mass spectrometry;hereditary metabolic diseases
串聯质谱检测技术(tandem mass spectrometry,MS/MS)是一种特异性的生化代谢物测定技术,通过测定物质的质荷比及相应离子强度,对物质进行定性和定量分析。 MS/MS技术能对干血滤纸片上微量血一次进行数十种氨基酸、有机酸、脂肪酸代谢异常的遗传代谢性疾病以及尿素循环障碍疾病进行检测[1],MS/MS超敏、高特异、快速,已广泛应用于遗传代谢病筛查,本研究用串联质谱技术对3228例样本进行检测,结合实验室其他指标和临床表现,效果满意,现报告如下。
1 资料与方法1.1 临床资料 检测对象为2018年4月~2019年5月在桂林市妇女儿童医院出生的新生儿和门诊或者住院部疑似遗传代谢病患儿共3228例,疑似患儿均有程度不等的喂养困难、反复呕吐、抽搐、黄疸、肝脾肿大、肌张力异常、精神运动发育迟缓、低血糖、高血氨、代谢性酸中毒、肝功能损害等。其中新生儿疾病筛查3084例(男175,例,女1329例),疑似遗传代谢病患儿144例(男85例,女59例)。3228例患儿年龄分布:2天~1个月 3084例,~3个月 31例,~6个月 22例,~1岁 23例,~12岁68例。
1.2 研究方法
1.2.1 标本采集 出生的新生儿满48 h并充分哺乳8次以上,采集足跟血制成干滤血片,共采集4个血斑,每个血斑直径≥8 mm,将血片悬空平置并常温下充分阴干,统一送到新生儿疾病筛查中心备检。疑似遗传病患儿采血方法和送检同上。均在家属知情同意下进行样本采集和检测。
1.2.2 串联质谱技术检测 采用AB Sciex Pte.Ltd公司的API 3200MD串联质谱仪器,以及PE公司生产的非衍生化多种氨基酸、肉碱和琥珀酰丙酮测定试剂盒。筛查指标共43 个,包括琥珀酰丙酮、11 种氨基酸、31 种酰基肉碱及相应比值,对28种遗传代谢病进行检测。试剂盒内供有一套用于测量琥珀酰丙酮、11种氨基酸的全套内标准品,另有一套用于测量肉碱类的13个内标准品和质控品。
1.2.3 检验原理 使用NeoBase测试对于氨基酸、琥珀酰丙酮、游离肉碱和酰基肉碱的测量过程包括使用含有内标准品的溶液对血片进行萃取以及使用串联质谱系统进行分析,每种分析物相对于内标准品的响应程度与它的浓度成比例。非衍生化多种氨基酸、肉碱和琥珀酰丙酮测定数据盒通过多反应监控(MRM)模式获取数据,在获取数据过程中,每种分析物碰撞诱导后的产物在设定时间内被测量,数据采集与处理由系统内软件包执行。每份报告单由遗传专科医师审核后发放。
1.2.4 可疑阳性患儿召回复查和诊断 筛查阳性患儿立即召回复查,根据指标超出截断范围的程度,结合临床表现,除了复查串联质谱,还要加做血尿常规、血糖、肝功能、血氨、血气分析、乳酸,脑电图、头颅CT等检查,高度怀疑某种遗传代谢病,再做基因检测确诊。确诊的患儿由遗传专科医师建立档案进行专案管理,定期追踪、随访、复查。
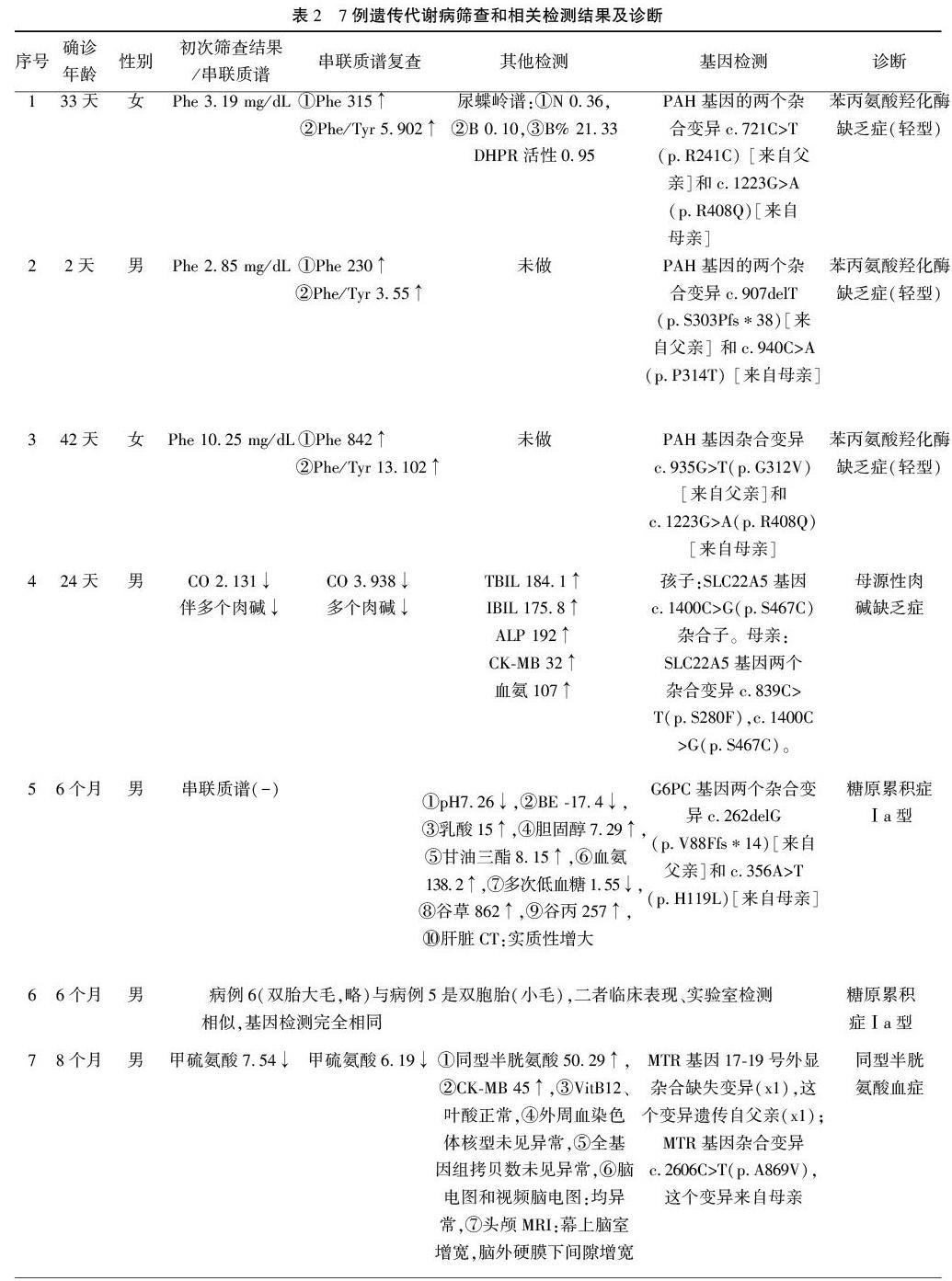
2 结 果2.1 阳性患儿检出和确诊情况 3228例中,可疑阳性171例,筛查阳性率5.30%,实际召回152例,召回率88.9%,最终确诊遗传代谢病7例(2.17‰)。分别是苯丙氨酸羟化酶缺乏症3例(0.93‰),母源性肉碱缺乏症1例(0.31‰),同型半胱氨酸血症1例(0.31‰),糖原累积症2例(0.62‰),其临床表现和实验室检测结果见表1、表2。
2.2 7例遗传代谢病患者临床转归 3例苯丙氨酸羟化酶缺乏症和1例母源性肉碱缺乏由于早期筛查、早期诊断、及时干预治疗效果显著,各项指标正常,经遗传专科医师建档管理、定期复查、指导饮食和药物治疗,生长发育和智力等与同龄儿相当。2例糖原累积症双胞胎患者半岁左右确诊后干预,各项生化指标趋于正常,但遗留智力和体格运动发育落后,肝大难恢复等不适。1例同型半胱氨酸血症生后8个多月确诊后干预,虽然各项生化指标正常,但仍存在喂养困难,间歇性抽搐未能控制,智力和体格发育落后等。
3 讨 论 遗传代谢病又称先天性代谢异常(inborn errors or metabolism,IEM或inherited metabolic diseases,IMD),是指有异常生化代谢标志物的一大类疾病,IEM因基因突变使合成的酶、受体、载体等蛋白功能缺陷,导致体内生化物质在合成、代谢、转运、储存等方面出现异常,包括氨基酸、有机酸、碳水化合物、脂肪酸等代谢紊乱和溶酶体、线粒体等细胞器内积聚、贮存异常而产生一系列临床症状的一大类疾病。遗传代谢病临床表现复杂多样、轻重不等、体内任何器官和系统均可受累,若得不到及时诊治,常导致智力低下、残疾甚至危及生命。常见有500~600种,虽然单一病种的患病率较低,仅几千分之一到几万分之一,但将IEM所有种类相加,其总体发病率可以高达0.5%以上[1]。本研究遗传代谢病总检出率2.71‰,远高于裴薇等人[2]报道的山东潍坊的检出率0.41‰,远低于赖光锐等[3]报道的沈阳的检出率1.83%和樊森等人[4]报道的南京的检出率2.5%,分析原因与样本的数量和来源有很大关系,山东潍坊的样本来源全部是新生儿,样本量7万多;沈阳和南京的样本来源全部是高危儿,样本量数千。在疾病种类方面,也由于样本量偏少未能覆盖有机酸代谢异常和尿素循环障碍疾病。氨基酸代谢性病中苯丙氨酸代谢异常检出率较高,且根据苯丙氨酸的值和基因诊断,均不属于经典型苯丙酮尿症,与杨莹等人[5]报道的贵州的检出均为经典型不同。
遗传代谢病提倡早期筛查,本组7例当中4例是在新生儿期自愿接受筛查的,全部得到及时的诊断和治疗,大部分代谢性疾病通过特殊的饮食控制,辅助适当药物治疗,花费不大,效果显著。病例1、2、3诊断为苯丙氨酸羟化酶缺乏症,属于氨基酸代谢病,假如没有早期筛查出来,会在3个月以后逐渐出现小便鼠尿味、喂养困难、体格和运动发育迟缓、毛发颜色变浅,甚至抽搐等神经系统并发症。病例4诊断为母源性肉碱缺乏症,属于脂肪酸β氧化障碍病,假如没有早期筛查出来,患者持续喂母乳,哺乳期间数个月后出现急性能量代谢障碍例如低血糖、高血氨、代谢性酸中毒;肝大、肝功能损害;严重者出现心肌病,表现为心室肥厚、心功能不全、肌酸激酶升高等[1]。这4个病例若等到发病时才被诊断,即使临床抢救治疗,效果都不理想。提醒临床医师早期筛查、早期诊治的重要性。糖原累积症属于碳水化合物代谢病,由于葡萄糖-6-磷酸酶(G6PC)先天性缺陷,导致低血糖、高乳酸血症,长期高乳酸血症导致生长迟缓;另一方面低血糖使脂肪大量动员,导致高脂血、脂肪肝;同时机体有机酸增多导致高尿酸血症。同型半胱氨酸血症也是氨基酸代谢病,半胱氨酸是多功能损伤因子,导致细胞结构和功能损伤,出生后数月出现呕吐、喂养困难、抽搐、肌张力低下、发育延迟等[1]。病例5、6、7早期没有进行筛查,都是在生后数个月出现上述症状体征在PICU住院时诊断出来,机体及各脏器发生了不可逆损害,治疗效果很有限。
基因检测可以助力遗传代谢病的确诊。近几年随着高通量测序技术的突飞猛进,对遗传病致病基因的诊断更加精准。本研究的7个确诊病例全部找到致病基因,且对查出致病位点患者进行家系验证寻找到致病来源,有利于优生优育。本组有6例患者查出父母都是携带者,这6例都是常染色体隐性遗传病,可根据常隐性疾病发生规律来指导夫妇怎样生育健康下一胎。病例4刚开始考虑原发性肉碱缺乏症,第一次进行的相关检测结果与患者临床不相符,由于游离肉碱能通过胎盘从母体转运到胎儿,如果母亲是患者,则胎儿在宫内肉碱供应不足及母乳中肉碱缺乏,致使婴儿出生后暂时性的母源性肉碱缺乏而被误诊,带着这样的思路我们再对母亲进行肉碱缺乏症相关基因检测,最终找到致病位点。假如没有基因检测加上临床经验不足势必会误诊,导致治疗策略有偏差。确诊后对母子同时治疗,并对母亲的胞弟(未婚)进行了串联质谱筛查,结果未见异常,对胞弟也进行了优生优育指导。
本研究例数较少,样本来源以本市区为主,还不能很好地反映桂林市单个遗传代谢病的发病率、病种构成比等,各种遗传代谢病的发生具有一定的地域性[6],大样本的筛查才能摸索出本地区遗传代谢病的发病特点,更好地应用遗传学原理指导优生优育,提高出生人口素质。
參 考 文 献
[1] 顾学范.临床遗传代谢病[M].北京:人民卫生出版社,2015:1-137.
[2] 裴薇,鹿相花,田维兵,等.潍坊地区新生儿遗传代谢病串联质谱筛查结果分析[J].中国中西医结合儿科学,2018,10(5):452-455.
[3] 赖光锐,李珍,张碧君,等.4819例遗传代谢病检测结果分析[J].国际遗传学杂志,2017,40(4):192-197.
[4] 樊森,张霞,黄艳军,等.遗传代谢病高危儿串联质谱及气相色谱筛查结果分析[J].东南大学学报(医学版),2018,37(6):1062-1065.
[5] 杨莹,平静,高莉蓉.4528例儿童血串联质谱检测遗传代谢性疾病结果分析[J].贵州医药,2014,38(5):445-447.
[6] 吕金峰,王伟青,李文杰.青岛市280例高危新生儿遗传代谢性病例分析[J].临床与病例杂志,2019,39(3):597-602.
