鱼鳔类肝素的分离纯化与结构鉴定
2019-08-30周斯仪钟赛意苏伟明杜振兴陈建平洪鹏志章超桦
周斯仪,钟赛意,2,3,*,苏伟明,3,杜振兴,陈建平,洪鹏志,章超桦,3,4
(1.广东海洋大学深圳研究院,广东 深圳 518108;2.广东海洋大学食品科技学院,广东 湛江 524088;3.广东省水产品加工与安全重点实验室,广东 湛江 524088;4.广东省海洋生物制品工程实验室,广东 湛江 524088)
鱼鳔也称鱼泡、鱼肚或鱼胶等,是鱼类调节沉浮的器官。鱼鳔富含胶原蛋白、多糖等活性物质,自北魏《齐民要术》开始记载以来,一直被视为药食两用的滋补珍品。《本草纲目》记载鱼鳔有止血作用,可用来治疗吐血、崩漏、外伤出血和产后抽搐等[1]。《本草新编》称鱼鳔具有养肝益肾、补肾固精的功效[2]。鱼鳔还能滋养经脉、散瘀消肿[3]。但其作用功效和物质基础仍不明确。目前对鱼鳔功能因子研究主要集中在其胶原蛋白和多肽的提取、结构表征及生物活性等方面[4-6]。近几年有报道发现鱼鳔多糖具有预防结肠癌、缓解胃溃疡、系统性红斑狼疮炎症的潜力[7-9],但目前对鱼鳔多糖的组成和结构特性尚未明确。本课题组前期研究发现鱼鳔多糖以糖胺聚糖为主[10],并进一步发现其主要由类肝素化合物组成。
肝素是由糖醛酸与葡萄糖胺组成的聚阴离子黏多糖,作为高效的抗凝血药物广泛应用于临床治疗。随着药理学的发展,发现肝素还具有抗血栓、抑制平滑肌细胞增生、抗炎症和抗肿瘤等生物活性,但其较高的抗凝血作用容易引起出血副作用,因而限制了其在非抗凝方面的应用[11]。类肝素是结构上与肝素结构相似的一类酸性黏多糖,常见的有硫酸乙酰肝素(heparan sulfate,HS)、硫酸软骨素(chondroitin sulfate,CS)和硫酸皮肤素(dermatan sulfate,DS)等。类肝素抗凝血作用相对较低,长期使用无肝素抗凝血活性引起的副作用。因此类肝素化合物越来越多地引起研究者关注。本研究采用酶法提取鱼鳔类肝素(heparinoid from swimming bladder,HSB),并对其进行分离纯化和结构鉴定,以期为阐明鱼鳔多糖的结构组成和构效关系提供参考,为鱼鳔功效因子的进一步开发与利用提供依据。
1 材料与方法
1.1 材料与试剂
鱼鳔(鲈鱼,Lateolabrax japonicus)购自湛江市霞山水产品批发市场;2709碱性蛋白酶(1.2×105U/g)南宁庞博生物工程有限公司;FPA98 Cl型大孔阴离子交换树脂 罗门哈斯(中国)公司;1-苯基-3-甲基-5-吡唑啉酮(5-methyl-2-phenyl-2,4-dihydro-3H-pyrazol-3-one,PMP)、CS二糖标准品(ΔDi-0S、ΔDi-UA-2S、ΔDi-4S和ΔDi-6S) 美国Sigma公司;乙腈(色谱纯)德国Meker公司;其他试剂均为国产分析纯。
1.2 仪器与设备
N-1300D-WB型旋转蒸发仪 东京理化器械株式会社;HR/T20MM型高速冷冻离心机 湖南赫西仪器装备有限公司;FD8508型真空冷冻干燥机 韩国ilShin公司;Xevo G2-XS QTof高分辨质谱(mass spectrometry,MS)仪 美国Waters公司;Agilent1200高效液相色谱(high performance liquid chromatography,HPLC)系统 美国安捷伦公司;Bruker AV-500和AV-700超导核磁共振(nuclear magnetic resonance,NMR)仪 德国Bruker BioSpin GmbH公司。
1.3 方法
1.3.1 HSB的制备与分离
参考文献[11]中肝素的制备工艺并略作调整,工艺流程为:鱼鳔干粉→酶解→灭酶→离心取上清液→大孔阴离子交换树脂吸附洗脱→醇沉→离心收集沉淀→无水乙醇洗涤2 次→透析→冷冻干燥。
具体步骤:鱼鳔经烘干、粉碎后,加入20 倍体积的双蒸水和质量浓度为5.4 mg/mL的蛋白酶,50 ℃水浴锅中酶解20 h;沸水浴灭酶10 min,冷却后离心(8 000 r/min、20 min);取上清液用大孔阴离子交换树脂进行动态吸附,再分别用0.3、0.5、0.9、1.1 mol/L氯化钠溶液进行梯度洗脱;每分钟收集一管,用阿利新蓝法分析管中类肝素含量,以收集管数为横坐标、阿利新蓝法染色所得吸光度(A490nm)为纵坐标制作梯度洗脱曲线;收集洗脱液,缓慢加入1 倍体积的无水乙醇,静置12 h后离心(4 000 r/min、5 min),收集沉淀,沉淀用无水乙醇洗涤2 次;经3 kDa透析袋脱盐,冷冻干燥。
1.3.2 基本成分质量分数和HSB得率的测定
类肝素质量分数测定采用阿利新蓝法[12],以肝素为标准品;蛋白质量分数测定采用福林-酚法[13],以牛血清白蛋白为标准品;糖醛酸质量分数测定采用咔唑-硫酸法[14],以葡萄糖醛酸为标准品;氨基己糖质量分数测定采用改进的Wagner法[15],以葡萄糖胺为标准品;硫酸基质量分数测定采用BaCl2-gel比浊法[16],以硫酸钾为标准品。HSB得率和各成分质量分数分别按式(1)、(2)计算。
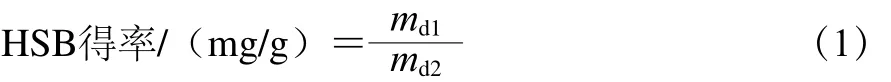
式中:md1为HSB质量/mg;md2为鱼鳔干粉质量/g。
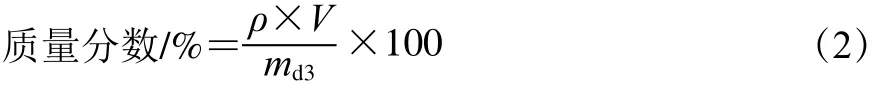
式中:ρ为某成分质量浓度/(mg/mL);md3为所称取HSB质量/mg;V为溶液体积/mL。
1.3.3 HSB的种类鉴定
类肝素化合物的结构鉴定采用醋酸纤维素薄膜电泳法,具体步骤参考文献[17]。
1.3.4 HSB的纯度分析
HSB用蒸馏水配成1 mg/mL的溶液,以蒸馏水为调零管,采用紫外-可见分光光度计测定样品的紫外吸收光谱。
1.3.5 HSB的分子质量分析
采用高效凝胶色谱(high performance gel permeation chromatography,HPGPC)测定HSB的分子质量[18]。色谱柱:Ultrahydrogel Column 500糖分析凝胶柱(7.8 mm×300 mm,10 μm);柱温:35 ℃;流动相:0.2 mol/L硫酸钠;流速:0.6 mL/min;检测器为1200示差检测器;进样体积:10 μL。
葡聚糖标准品(分子质量分别为10 000、23 800、48 600、80 000、150 000、273 000、409 800 u)和HSB分别用0.2 mol/L硫酸钠溶液配制成质量浓度为5 mg/mL的溶液,经0.22 μm的针式滤膜过滤后进样。记录标准品和样品的保留时间并进行数据处理。
1.3.6 HSB的单糖组成分析
样品前处理:称取样品3.0 mg于15 mL样品瓶中,加入1.5 mol/L的三氟乙酸9 mL,置于110 ℃烘箱中分别水解2、4、6、8、12、16、24、28 h,水解液用氮气吹干,蒸干物用超纯水溶解,并于-20 ℃下保存备用。
处理后的样品和单糖标准品采用PMP衍生-高效液相反相色谱法测定[19]。单糖标准品:甘露糖(mannose,Man)、鼠李糖(rhamnose,Rha)、葡萄糖醛酸(glucuronic acid,GlcA)、艾杜糖醛酸(iduronic acid,IdoA)、N-乙酰半乳糖胺(N-acetylgalactosamine,GalNAc)、半乳糖(galactose,Gal)。色谱柱:ZORBAX EclipseXDB-C18色谱柱(4.6 mm×250 mm,5 μm);柱温:30 ℃;流动相:0.02 mol/L磷酸盐缓冲液(pH 6.0)-乙腈溶液(83∶17,V/V);流速:1.0 mL/min;检测波长:245 nm;进样体积:10 μL。
1.3.7 HSB结构的FTIR分析
取2.0 mg样品,在红外灯下烘烤2 h,然后放入玛瑙研鉢中,加入100 倍质量的以同样方式处理的氯化钾,混合均匀,研磨至颗粒小于2.5 μm,放入压片机中加压制成半透明的小薄片;再将其放入傅里叶变换红外光谱(Fourier transform infrared spectrophotometer,FTIR)扫描仪中进行光谱扫描,扫描范围为4 000~400 cm-1。
1.3.8 HSB的二糖组成分析
二糖组成分析参考文献[20]的方法并稍作修改。精密称取0.010 0 g多糖样品,加入5 mL醋酸铵缓冲液(pH 7.6~8.0,含0.2 U CS酶ABC),37 ℃孵育24 h。沸水浴灭酶5 min,10 000 r/min离心25 min。上清液用3 kDa超滤离心管过滤,取滤液冷冻干燥。将处理后的样品和CS二糖标准品(ΔDi-0S、ΔDi-UA-2S、ΔDi-4S和ΔDi-6S)分别用超纯水配成1 μg/mL溶液用于MS/MS分析。
MS条件:电子轰击离子源;电子能量70 eV;传输线温度275 ℃;离子源温度200 ℃;母离子m/z 285;激活电压1.5 V;质量扫描范围m/z 35~500。
1.3.9 HSB结构的NMR分析
HSB样品溶于D2O中,冷冻干燥,反复3 次以置换出H2O。处理后的样品用D2O配制成30 mg/mL的溶液,在常温下用NMR仪进行1H谱、13C谱、异核单量子相干(heteronuclear singleqauntum coherence,HSQC)谱和异核多键相关(heteronuclear multiple-bond correlation,HMBC)谱分析。
1.4 数据分析
所有实验重复3 次,结果以平均值±标准偏差表示,采用Origin 9.1和ChemBioDraw Ultra软件作图。
2 结果与分析
2.1 HSB的分离结果
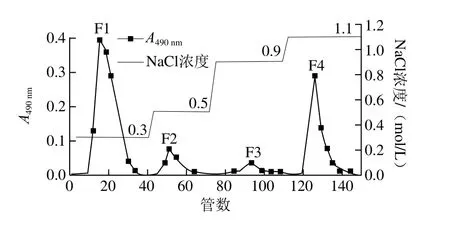
图1 类肝素化合物FPA98 Cl型大孔阴离子交换柱洗脱曲线Fig. 1 Elution curve of heparinoids on FPA98 Cl column
类肝素为聚阴离子酸性黏多糖,在组织中以糖蛋白的形式存在。因此,类肝素的提取以酶法为主,蛋白酶将糖蛋白中的蛋白质分解成易于去除的小肽和氨基酸,使类肝素从中释放出来。游离在酶解液中的类肝素经FPA98 Cl型大孔阴离子交换树脂动态吸附,再用氯化钠溶液梯度洗脱,使中性多糖、蛋白质和多肽等杂质被低浓度的氯化钠溶液洗脱下来,带阴离子基团较多的类肝素则被高浓度氯化钠溶液洗脱下来。梯度洗脱结果图1所示,0.3、0.5、0.9、1.1 mol/L氯化钠溶液分离出的洗脱液经醇沉、透析、冷冻干燥后,得到4 个组分:F1、F2、F3和F4。各洗脱组分的得率和基本成分质量分数见表1。
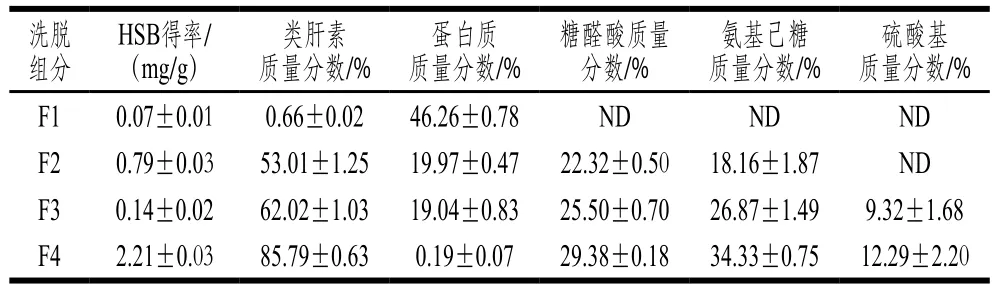
表1 各洗脱组分的HSB得率与基本成分Table 1 Yield and composition of each elution fraction
由表1可知,从组分F1到F4,类肝素、糖醛酸和氨基己糖质量分数逐渐升高,蛋白质量分数递减,说明该方法能将类肝素高效分离出来。组分F4的HSB得率最高,为(2.21±0.03)mg/g,F2次之;组分F1蛋白质质量分数最高,HSB得率和类肝素质量分数都很低,且检测不到糖醛酸和氨基己糖,说明组分F1可能为中性糖和蛋白质的混合物,梯度洗脱曲线中该组分吸光度高可能受色素、蛋白质等杂质的影响。组分F4的硫酸基质量分数最高,为(12.29±2.20)%,F3次之,其他组分未检测到硫酸基。硫酸基质量分数是提示类肝素活性的一项重要指标,一定范围内硫酸基的质量分数越高,生物活性越强[21]。Sayari等[17]用海鳌虾壳提取的类肝素中硫酸基质量分数高达(23.00±0.03)%,具有较强的抗凝血和抗细胞增殖作用。
2.2 HSB的种类鉴定结果
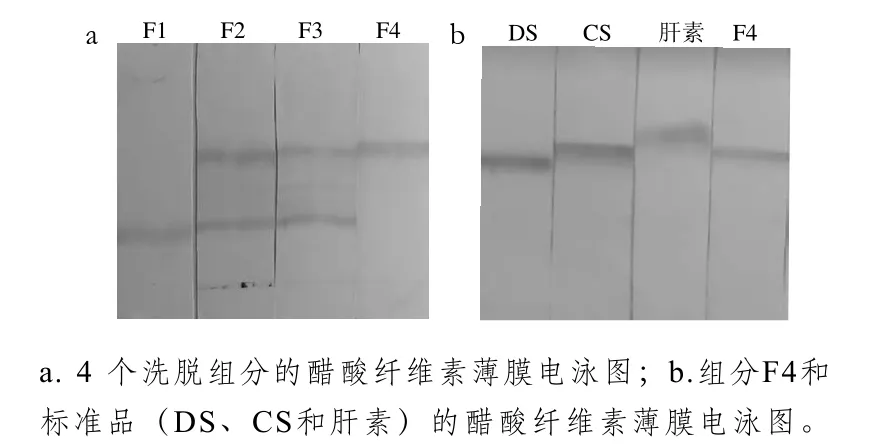
图2 各洗脱组分及标准品的醋酸纤维素薄膜电泳结果Fig. 2 Acetate cellulose electrophoresis of each elution fraction and glycosaminoglycan standard
不同种类的糖胺聚糖之间存在电荷密度差异,在醋酸纤维素薄膜电泳中表现出不同的迁移率。各洗脱组分醋酸纤维素薄膜电泳结果如图2a所示,组分F1为单一条带,迁移率最低,这与其基本成分分析相符,进一步验证F1可能是中性糖;组分F2和F3有4 个条带,说明所含糖胺聚糖种类较多;组分F4为单一条带,迁移率最大。结合各组分HSB得率和理化性质分析,选择类肝素、糖醛酸、氨基己糖和硫酸基质量分数最高、蛋白质量分数低、醋酸纤维素薄膜电泳条带单一的组分F4做进一步结构鉴定。
醋酸纤维素薄膜电泳操作简易、灵敏度高,可作为初步鉴别糖胺聚糖种类的方法。在吡啶-甲酸缓冲液(pH 3.0)中,相对迁移率从小到大依次为透明质酸、硫酸角质素、DS、CS和肝素[20]。由图2b可知,肝素因电荷密度最高,迁移率最大;其次分别是CS、DS。组分F4的条带与CS迁移率相近,初步确认组分F4为CS。
2.3 组分F4的纯度及分子质量
2.3.1 紫外光谱扫描结果
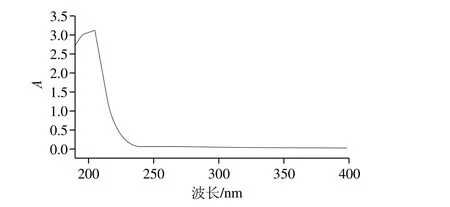
图3 组分F4紫外光谱图Fig. 3 Ultraviolet absorption spectrum of F4
紫外光谱扫描是评价类肝素纯度的方法之一。类肝素在185~220 nm处有糖基末端的吸收峰[8],核酸和蛋白质分别在260、280 nm处有特征吸收峰。由图3可知,紫外光谱扫描结果显示,组分F4除在204 nm处有糖基末端吸收峰外,在其他紫外区域无明显吸收峰;结合2.1节组分F4的蛋白质量分数为(0.19±0.07)%,表明F4纯度较高。
2.3.2 HPGPC分析结果
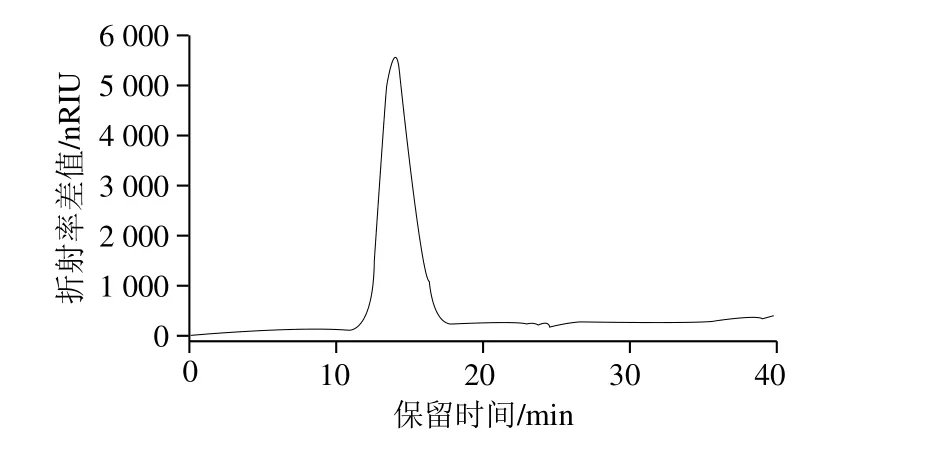
图4 组分F4的HPGPC图谱Fig. 4 High performance gel permeation chromatogram of F4
由图4可知,组分F4的HPGPC图只出现1 个峰,且峰形较对称,表明F4的质量分布相对均一,纯度较高。高效凝胶色谱与示差折光检测器联用测得葡聚糖的分子质量的对数(y)与保留时间(x)的回归方程为:y=-0.319 2x+9.429 0(R2=0.996 6),计算得出组分F4的分子质量约为84 000 u。一般类肝素的分子质量在5 000~40 000 u范围之间[22],相比之下,组分F4的分子质量较大。
2.4 组分F4的单糖组成分析结果
糖胺聚糖由特定的重复二糖单位构成,根据二糖组成的差异,分为透明质酸、HS、肝素、DS和CS。因此,可以根据单糖组成推测类肝素黏多糖的种类。对比单糖混标的HPLC图谱(图5)发现,组分F4单糖衍生物(水解时间8 h)的HPLC图(图6)中出现1 个峰值很高的未知峰。由图7可知,随着组分F4水解时间的延长,未知峰的峰面积比例减少,GlcA和IdoA的峰面积比例也逐渐减少,GalNAc的峰面积比例不断升高,说明未知峰可能是未降解的寡糖片段。另外,也有报道称此处为未降解完全的寡糖片段[23]。这可能是由于半乳糖胺较难水解,有研究表明半乳糖胺比其他单糖需要更长的时间才能完全被降解[19,24]。糖醛酸在酸溶液中不稳定,易发生脱羧反应,甚至会被强酸完全分解[25],因此随降解时间的延长糖醛酸的峰面积比例降低。根据保留时间和峰面积比例得出,组分F4主要含GlcA、GalNAc和少量的IdoA、Gal,由此得出其主链可能是CS。
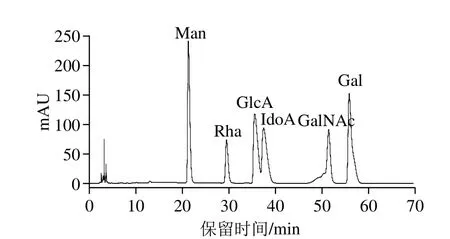
图5 混合标准品单糖衍生物的HPLC图谱Fig. 5 High performance liquid chromatogram of mixture of monosaccharide derivative standards
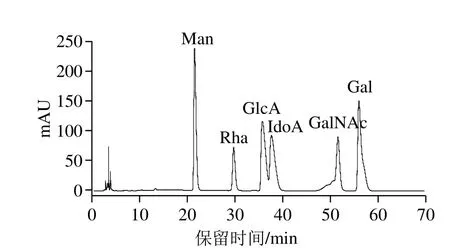
图6 组分F4单糖衍生物的HPLC图谱Fig. 6 High performance liquid chromatogram of monosaccharide derivatives from F4
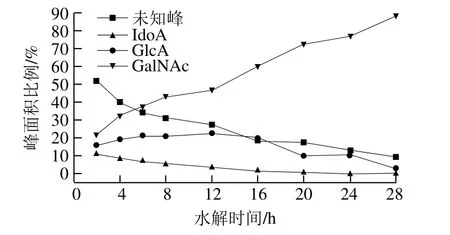
图7 水解时间对组分F4降解产物峰面积比例的影响Fig. 7 Effect of hydrolysis time on percentage peak areas of degradation products from F4
2.5 组分F4结构的FTIR分析结果
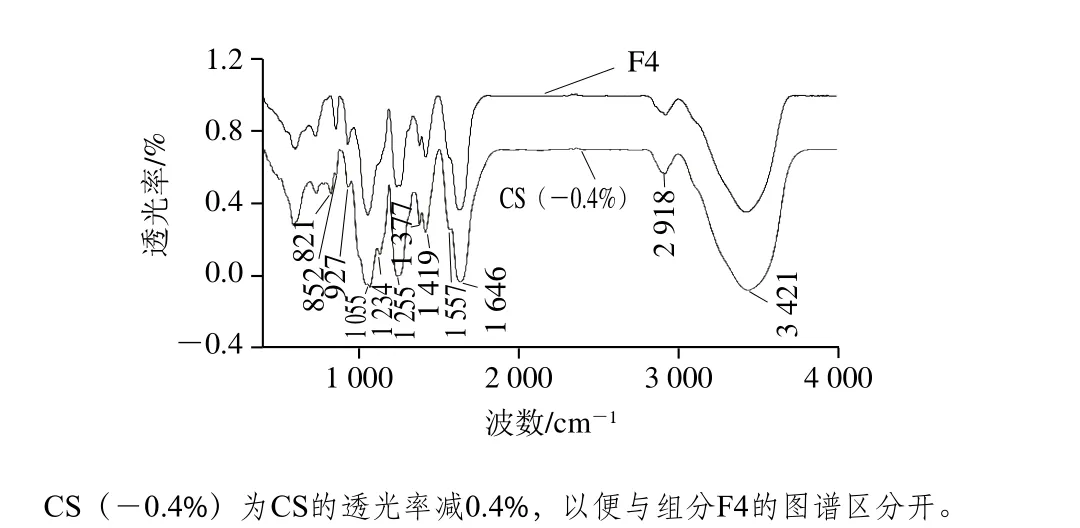
图8 组分F4和CS的FTIR图Fig. 8 Fourier transform infrared spectra of both F4 and CS
由图8可知,组分F4的FTIR图与CS标准品基本一致。3 421 cm-1处出现强而宽的峰是O—H和N—H伸缩振动,说明组分F4存在分子间和分子内的氢键[26];2 918 cm-1处的吸收峰表明存在亚甲基[27];1 646 cm-1处强而稍宽的峰是乙酰氨基中的C=O对称伸缩振动产生的,另外,1 557 cm-1处N—H变角振动峰和1 419 cm-1处C—N伸缩振动峰表明存在乙酰氨基[28-29];1 377 cm-1处的峰是—COO-中的C=O对称伸缩振动产生的[27];1 234 cm-1处不太尖锐的峰可能是C—H变角振动;1 255 cm-1处硫酸基中的S=O伸缩振动和852 cm-1处硫酸酯基中的C—O—S轴向伸缩振动表明存在硫酸基团[35];927 cm-1处的吸收峰是吡喃糖环的非对称伸缩振动产生的[30]。
根据二糖单位中硫酸基数量和链接位置的不同,CS分为A、C、D、E等种类。A型CS(CSA)的硫酸基在C4位,产生的轴向伸缩振动峰在850 cm-1附近;C型CS(CSC)的硫酸基在C6位,硫酸基处于平位,吸收峰在820 cm-1附近[31]。本实验所用CS标准品为E型CS(CSE),在820 cm-1和850 cm-1附近均有吸收峰。组分F4只在850 cm-1附近有吸收峰,表明F4可能为CSA或其衍生物。
2.6 组分F4的二糖组成分析结果
根据以上结果得出组分F4中主要含CS。为进一步分析组分F4的二糖组成,采用CS酶ABC将CS完全裂解成不饱和二糖,再经MS/MS分析鉴定。根据硫酸基团数量及链接位置,CS二糖分为ΔDi-0S、ΔDi-UA-2S、ΔDi-4S和ΔDi-6S等。其中,ΔDi-4S是CSA的主要二糖单位,ΔDi-6S是CSC的主要二糖单位。此外,由于具有相同硫酸基团数量的二糖相对分子质量相同,不能通过准离子峰区分开,因此通过二级质谱的碎片离子进行区分。组分F4完全降解产物和4 种常见的CS二糖标准品的MS/MS结果如图9所示。
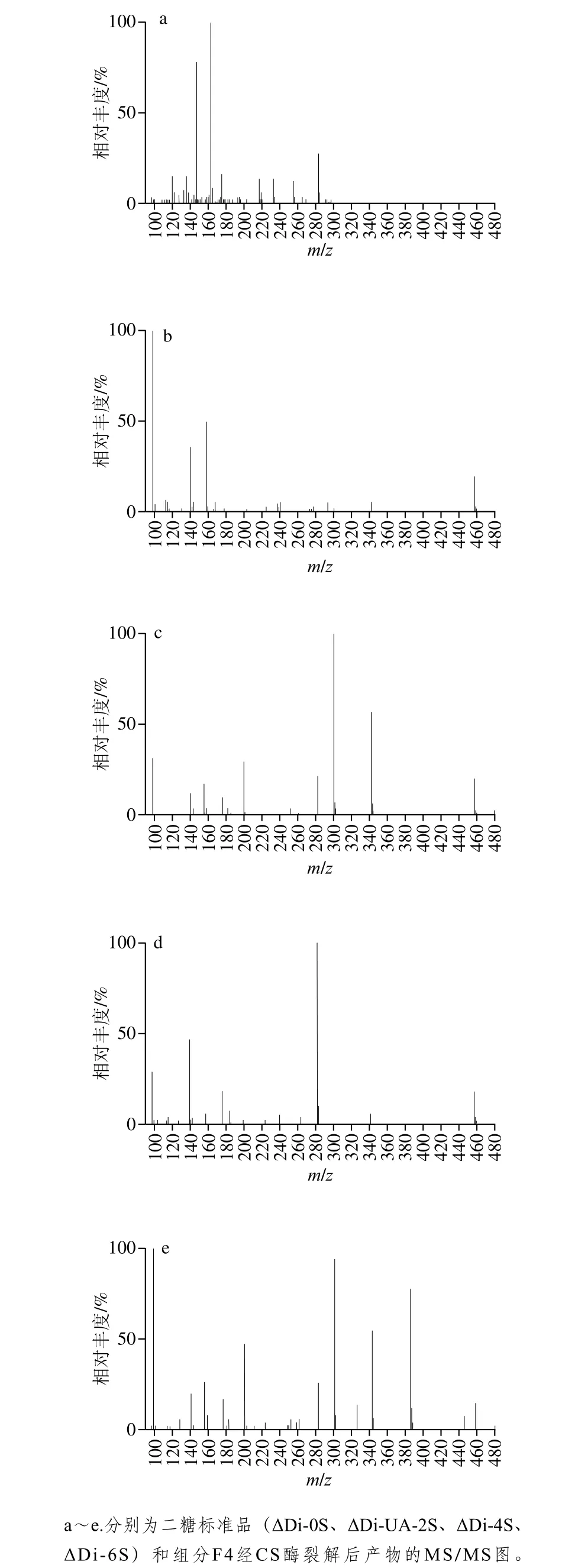
图9 组分F4裂解产物和二糖标准品的MS/MS图Fig. 9 MS/MS spectra of degraded products of F4 and disaccharide standards
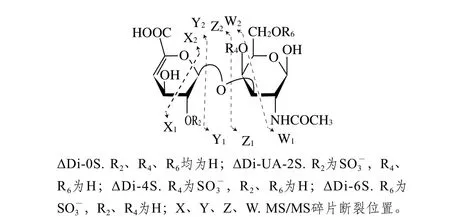
图10 CS二糖碎片基本组成图[32-33]Fig. 10 Basic composition of chondroitin sulfate disaccharide fragments[32-33]
图10 为CS二糖碎片的基本组成[32-33]。结合图10和表2可知,ΔDi-0S和ΔDi-UA-2S的半乳糖胺不含硫酸基,特征碎片结构是[Z1-2H]-,m/z为202.09;ΔDi-UA-2S的硫酸基连接在葡萄糖醛酸C2的羟基上,因此具有[Y2-H]-和[Z2+Na-H]-特征碎片结构,其m/z分别为236.99、276.98。ΔDi-4S和ΔDi-6S的区别体现在,ΔDi-4S在m/z282处的离子相对丰度小于m/z300或不存在,而ΔDi-6S在m/z300处的离子相对丰度小于m/z282或不存在[34]。图9中ΔDi-4S在m/z300.06处的离子相对丰度大于m/z282.05,ΔDi-6S在m/z282.05处的离子相对丰度较大,在m/z300.06处没有碎片峰。此外,还观察到ΔDi-6S在m/z239.94处的特征峰,其结构为[W1-H]-,可用来判断是否存在ΔDi-6S。组分F4完全降解产物的质谱图中,在m/z202.09、236.99、276.98处没有碎片峰,表明组分F4不含ΔDi-0S和ΔDi-UA-2S;在m/z300.06处的离子相对丰度大于m/z282.05,且不含ΔDi-6S特征峰m/z239.94,说明F4的主要二糖单位为ΔDi-4S。结合FTIR扫描结果,确定组分F4主要为CSA。

表2 二糖MS/MS特征碎片峰及其结构Table 2 MS/MS characteristic fragment peaks of disaccharides and their structures of disaccharides
2.7 组分F4结构的NMR鉴定结果
图11为组分F4的1H、13C、HSQC和HMBC的NMR谱图。在1H NMR谱图中,质子信号最强的峰位于δ4.79处,是氘代水产生的溶剂峰,除此之外,所有质子信号均位于2 个光谱区。在δ2.0~2.1之间出现了乙酰胺甲基信号,文献[35]报道CSA和CSC的乙酰胺甲基信号分别在δ2.04、2.02处,组分F4乙酰胺甲基信号位于δ2.04,说明F4可能为CSA;其他质子信号集中在δ3~5之间,说明F4为β-构型[36]。

图11 组分F4的NMR 图谱Fig. 11 NMR spectraof F4
在13C NMR谱图中,δ174.96和δ174.35的低场信号表明存在乙酰氨基和己糖醛酸的羧基;δ22.45的高场信号可能为乙酰胺甲基碳;其他信号集中在δ50~105之间。此外,由HSQC谱图中的(2.04,22.47)和HMBC谱图(2.04,174.38)确定δ22.47为乙酰氨基的甲基碳信号,δ2.04为乙酰氨基的甲基质子信号,δ174.38为乙酰氨基的羰基碳信号。参照文献[35]和[37]对组分F4的1H、13C NMR谱进行归属,结果如表3所示。对1H、13C、HSQC和HMBC图谱进行综合分析,得出组分F4主要结构式为[→4GlcUA β1→3GalNAc(4S)β1→]n。
表3 组分F4的1、13C NMR信号归属Table 3 1E and 13C nuclear magnetic resonance signal assignment of F4

表3 组分F4的1、13C NMR信号归属Table 3 1E and 13C nuclear magnetic resonance signal assignment of F4
信号归属 1 2 3 4 C-5 6 NAc-1 NAc-2 GlcA-H 4.47 3.38 3.59 3.79 3.67 GlcA-C 103.39 72.07 73.34 80.19 76.27 174.96 GalNAc-H 4.57 4.03 4.02 4.75 3.84 3.79 2.04 GalNAc-C 100.65 51.25 75.35 76.23 74.23 60.73 174.38 22.47
3 结 论
本研究以鱼鳔为原料,采用酶法提取类肝素,通过阴离子交换树脂和醇沉进行分离纯化,制备出得率较高、结构相对均一的类肝素组分F4,其得率为(2.21±0.03)mg/g,类肝素质量分数为(85.79±0.63)%。紫外光谱和HPGPC结果表明组分F4纯度较高;分子质量分布相对集中,约为84 000 u。通过柱前衍生-HPLC得出组分F4主要含GlcA、GalNAc和少量的IdoA、Gal。组分F4的FTIR谱图和二糖组成与CSA一致,结合NMR分析得出组分F4主要结构式为[→4GlcUA β1→3GalNAc(4S)β1→]n,属于CSA。CSA具有免疫调节[38]、抗炎[39]、抗氧化[40]和抗肿瘤[41]等活性,主要用于预防骨关节炎、冠心病、神经痛及角膜炎等疾病[11]。本研究可为进一步分析鱼鳔功能及其作用机理、提高鱼鳔的利用价值提供参考。
