具有非典型影像改变的肺泡蛋白沉积症3例报道并文献复习
2019-08-24赵婷婷蔡后荣李燕李慧孙琦张英为
赵婷婷,蔡后荣,李燕,李慧,孙琦,张英为
肺泡蛋白沉积症(pulmonary alveolar proteinosis,PAP)是一种罕见的肺部疾病,在1958年被Rosen及其同事首次报道,其患病率大约4/100万[1]。胸部CT表现为“铺路石征”或“地图样”分布,结合支气管镜肺泡灌洗液(bronchoalveolar lavage fluid,BALF)呈现特征性的过碘酸雪夫(periodic acid schiff,PAS)染色阳性物质沉积即可确立诊断[2]。但当胸部CT表现不典型,BALF呈PAS阴性反应时,易出现误诊。现报道3例胸部CT表现不典型的PAP,并结合文献进行复习,提高临床医师对非典型PAP的认识。
1 临床资料
例1.男,53岁。因“体检发现肺间质改变5月”于2008年9月1日入院。患者5个月前体检时全胸片示肺间质改变。因无不适,未进一步诊治。1个月前复查胸片较前未见吸收,查胸部CT示双肺纵隔旁及胸膜下少许磨玻璃影及网状影(图1A、1B)而入院。从事铸造工作10年。入院查体:T 36.0℃,P 72次/min,R 20次/min,BP 110/64 mmHg。双肺未闻及干湿性啰音,心腹查体未见异常,双下肢无水肿。初步诊断:职业相关性间质性肺炎?入院后完善检查,血气分析(未吸氧):pH 7.424,PaO279 mmHg,PaCO238.4 mmHg;血常规、生化、免疫均正常,自身抗体、血抗中性粒细胞胞浆抗体(ANCA)均阴性;肺功能示轻度混合性通气功能障碍。为进一步明确诊断,9月4日行胸腔镜下肺活检。术后病理提示:多数肺泡内见大量粉染蛋白样物质充盈,颗粒状;特殊染色:PAS(+),PAS-D(+),AB(-),GMS(-)(图2A见封3)。明确诊断为肺泡蛋白沉积症。综合患者临床表现、血气分析及肺功能等,未予特殊处理,医嘱出院。门诊随访5年,病情稳定。后失访。
例2.男,37岁。因“反复咳嗽、咯痰1年余”于2014年11月12日入院。患者1年前开始出现咳嗽、咯痰,痰色白质稠,伴活动后气喘,无发热,抗感染治疗疗效不佳。2013年11月外院查胸部CT示两肺斑片影,密度不均。PET/CT示两肺多发片絮状及结节状高密度影,部分结节代谢增高。行电子支气管镜检查未见异常,临床考虑肺结核,予利福喷丁、乙胺丁醇、吡嗪酰胺等试验性抗结核治疗。治疗约9个月后症状未缓解,后至我院门诊复查胸部CT示:病灶较前无明显吸收(图1C、1D)。故入住院。个人史及家族史无特殊。入院查体:T 37.1℃,R 20次/min,P 66次/min,BP 129/73 mmHg。口唇无紫绀,两下肺可闻及捻发音,心腹查体未见异常,双下肢无水肿。初步诊断:间质性肺炎。
入院后完善检查,血常规、生化、血气分析、肺癌四项、免疫常规均正常;自身抗体、ANCA、痰涂片找抗酸杆菌均阴性;T细胞γ干扰素检查阳性。肺功能检查示中度混合性通气功能减退,弥散功能基本正常。为进一步明确诊断,2014年11月17日行胸腔镜下肺活检。术后病理示:送检肺组织见部分区域肺泡腔、远端气道内大量均质、嗜伊红样物质沉积,部分区域肺泡隔炎性增宽,组织学符合肺泡蛋白沉积症(图2B见封3)。予雾化吸入重组人粒细胞巨噬细胞刺激因子治疗(75 μg,每天2次,隔周应用)。后失访。
例3.女,48岁。因“反复咳嗽、气喘1年余,加重伴发热8 d”于2016年3月25日入院。患者1年前无明显诱因出现咳嗽,咯少许白黏痰,伴活动后气喘。2015年5月于当地医院查胸部CT示左下肺实变,两肺间质性炎性反应。行支气管镜检未见异常。1年来症状迁延,时轻时重。8 d前受凉后症状加重伴发热,体温最高38.6℃,当地医院治疗效果不佳,遂来我院。既往有甲状腺功能减退病史。入院查体:T 36.3℃,R 20次/min,P 67次/min,BP 122/72 mmHg。口唇无紫绀,双肺呼吸音粗,双下肺可闻及捻发音,余阴性。入院血气分析(鼻导管吸氧2 L/min):pH 7.441,PaO280 mmHg,PaCO239.9 mmHg。初步诊断:(1)间质性肺炎伴感染;(2)Ⅰ型呼吸衰竭;(3)甲状腺功能减退症。入院后检查,血WBC 22.3×109/L,N 80%,Hb 111 g/L。肺功能检查示中度以限制为主的混合性通气功能减退,弥散功能重度减退。胸部CT示两肺弥漫性磨玻璃影及网状影,部分斑片及实变影,伴小叶间隔增厚(图1E、1F)。为明确诊断,2016年4月5日行胸腔镜下肺活检。术后病理示:多数区域肺泡腔内见多少不等的嗜伊红蛋白样物质沉积,伴多量胆固醇结晶形成,组织学符合肺泡蛋白沉积症(图2C见封3)。予雾化吸入重组人粒细胞巨噬细胞刺激因子治疗(150 μg, 每天2次,隔周应用),病情改善后出院。2016年8月因症状加重伴发热再次入住我科,查血WBC 20.6×109/L,N 72.2%,Hb 77 g/L,PLT 65×109/L。遂于8月29日行骨髓穿刺检查,明确为多发性骨髓瘤(MDS-EB-2)。2016年9月19日开始行CAG方案化疗(阿柔比星12 mg,静脉注射,d 1~8,阿糖胞苷 0.015 g,皮下注射,q 12 h,d 1~14,重组人粒细胞刺激因子注射液 300 μg,皮下注射,d 1~14,白细胞<20×109/L用)。化疗期间症状改善不佳,2016年12月死于严重肺部感染。

注:A、B.例1 胸部HRCT示双肺纵隔旁及胸膜下少许磨玻璃影及网状影;C、D.例2 胸部HRCT示两肺网状阴影、磨玻璃结节影及部分实性结节影;E、F.例3 胸部HRCT示两肺弥漫性磨玻璃影及网状影,散在斑片影,伴小叶间隔增厚
图1 胸部HRCT检查影像学变化
2 文献回顾
先后以“非典型”并“肺泡蛋白沉积症(atypical pulmonary alveolar proteinosis)”以及“肺泡蛋白沉积症”并“误诊”为关键词在中国知网和万方数据库中检索,以“atypical pulmonary alveolar proteinosis”为检索词通过PubMed数据库进行检索,检索时间自1980年3月1日—2018年12月31日,并重点关注误诊病例的影像特征描述部分,剔除儿童病例,共检索到中文文献2篇,1篇为个案报道,1篇为临床总结分析;英文文献12篇,均为个案报道,见表1。
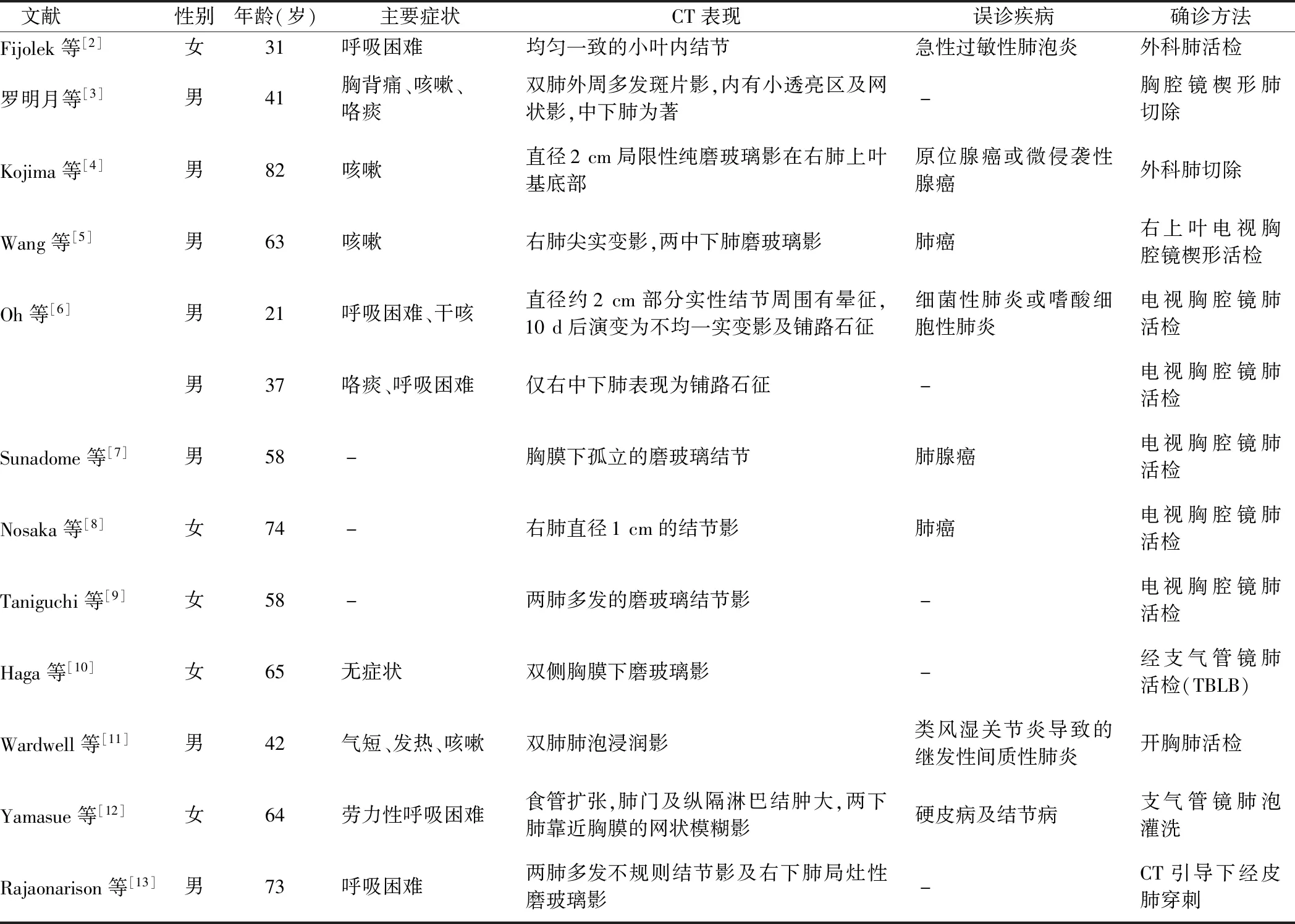
表1 非典型PAP临床资料比较
3 讨 论
肺泡蛋白沉积症(PAP)是一类以肺泡表面活性物质在肺泡腔的异常聚积为特点的疾病,其机制为肺泡巨噬细胞清除表面活性物质能力下降[14]。根据发病原因不同,PAP分为3类:(1)自身免疫性PAP,是由于粒细胞—巨噬细胞集落刺激因子(granulocyte macrophage colony stimulating factor,GM-CSF)自身抗体存在所致;(2)先天性PAP,由于编码表面活性物质基因突变所致;(3)继发性PAP,由于血液系统或实体恶性肿瘤、无机粉尘吸入、化疗、机会性感染、赖氨酸尿性蛋白质不耐受等引起[15]。
PAP往往起病隐匿,亚急性病程[16]。临床表现并无特异性,大约有1/3的患者无明显症状,咳嗽、气喘为较常见症状;部分患者可有杵状指及肺部湿啰音[14]。
胸部影像学检查是诊断PAP的重要手段。典型的胸部CT特点为“铺路石征”和“地图样”分布。当HRCT提示以上征象时,诊断PAP并不困难。但尚有PAP的影像学表现并非典型影像特征,根据文献报道这些表现包括:(1)结节影,包括部分实性结节[6]、孤立的纯磨玻璃结节[4,7]、两肺多发的磨玻璃结节[9]、均匀一致的小叶内结节[2]、部分钙化的结节。(2)磨玻璃影,除磨玻璃结节影外,尚有片状分布的磨玻璃影,但不与小叶间隔增厚重叠存在。(3)局限分布的“铺路石征”[6]。(4)网状阴影[3,12]、斑片及实变影[3,5-6]。(5)不同的影像学特点之间可以相互演变[6]。综上所述,当出现上述不典型征象时,较易出现误诊。PET/CT对于PAP的诊断并无明显帮助,甚至会增加诊断难度[5]。
特征性的HRCT表现以及典型BALF结果对于诊断PAP是足够的,不需要组织病理学检查,但对于影像学表现不典型的病例,仅靠典型的BALF诊断是不够的[17],此时还需要组织病理学确诊。本次报道的3例患者均经开胸肺活检确诊。文献报道的病例中,外科肺活检取材是主要手段。故当患者的影像学无明显特异性、支气管镜诊断难以明确时,应尽快行外科肺活检明确诊断。
先天性、特发性PAP或轻型的PAP可应用非侵袭性医疗策略,如皮下注射或雾化吸入GM-CSF、单克隆抗体、血浆置换、高压氧等。继发性或重型PAP需全肺灌洗[1]。全肺灌洗目前被认为是最有效的治疗方法,最终的治疗方法则是肺移植[1]。
综上所述,随着越来越多的PAP被诊断及报道,PAP的CT表现多种多样,已不局限于“铺路石征”或“地图样”分布。当患者胸部CT出现分布各异的磨玻璃影、结节影、实变影及网状阴影时,在鉴别诊断时需考虑到PAP的可能;对于BALF或TBLB诊断困难者,需尽快完善外科肺活检,以免延误诊治。
利益冲突:无
作者贡献声明
赵婷婷: 文献检索、总结及论文撰写;蔡后荣:文章修订;李燕、李慧:提供病例资料;孙琦:核对病理图片;张英为:提出研究思路及修订文章
