新生儿川崎病1例病例报告
2019-08-23任静王莹
任 静 王 莹
作者单位 苏州大学附属儿童医院新生儿科 苏州,215000
1 病例资料
男,24 d,因“发热3 h”入苏州大学附属儿童医院(我院)。3 h前无明显诱因出现发热,热峰38.5℃。稍有吐沫,打喷嚏,奶量下降,热时略烦躁,无抽搐,无咳嗽,无青紫。
患儿足月,因“瘢痕子宫”剖宫产娩出,出生史无异常提示。
入院查体:体温37.7℃,脉搏138·min-1,呼吸44·min-1,体重4.74 kg。神清,精神反应可。颜面部皮肤稍黄染并有少许散在皮疹,前囟平软。颈部触及数枚肿大淋巴结。双肺呼吸音粗,心律齐,心音可,心前区未及杂音。腹软,肝脾肋下未扪及。四肢肌张力可。肢端暖,末梢循环可。
实验室检查:血常规WBC 13.6×109·L-1,N 0.56, Hb 132 g·L-1,PLT 434×109·L-1,CRP 3.1 mg·L-1,降钙素原<0.25 ng·mL-1。血白蛋白37.4 g·L-1,ALT 10.3 U·L-1,总胆红素161 μmol·L-1,直接胆红素11.9 μmol·L-1。肾功能指标未见异常。红细胞沉降率(ESR) 42 mm·h-1。脑脊液和血培养阴性。流感、副流感和合胞病毒均阴性,支原体抗体、EV-71 IgM、TORCH、结核抗体和自身抗体均阴性。体液免疫指标IgA 0.05 g·L-1,IgG 4.16 g·L-1,IgM 0.21 g·L-1;细胞免疫指标CD3+63%,CD4+40.6%,CD8+20.6%,CD4+/CD8+2.0;CD19+CD23+11.5%。
影像学检查:颅脑和腹部B超未见异常,B超提示左侧颈部数枚肿大淋巴结,最大约15 mm×7 mm。X线胸片提示新生儿肺炎,肠郁张。颅脑CT提示白质密度略偏低。心脏彩超显示,房间隔缺损 3.1 mm;双侧冠脉未见扩张,左主干内径1.6 mm,右主干内径1.3 mm,未见扭曲,内膜线平滑,主动脉瓣环内径9.0 mm。
诊疗经过:入院后予拉氧头孢抗感染及支持治疗,入院当天体温降至正常。第2 d再次出现发热,热峰39℃,且开始出现皮疹,压之褪色,以手心、脚心、卡疤周围、肩部、前胸部为主(图1A、B)。第3 d皮疹继续扩展至背部、四肢及胸腹部,CRP 44.2 mg·L-1、降钙素原0.63 ng·mL-1,皮肤科会诊考虑多形红斑,改用头孢哌酮舒巴坦抗感染及甲泼尼龙1 mg·kg-1治疗,皮疹及发热无明显改善(图1C);改用美罗培南(40 mg·kg-1),甲泼尼龙加至2 mg·kg-1,并予IVIG 800 mg·kg-11次,患儿体温逐渐下降(肛温36.4~37.6℃),皮疹较前消退,出现口唇皲裂(图1D)。第7 d再次出现发热,复查血常规PLT 762×109· L-1,B超提示左侧颈部淋巴结肿大,考虑不完全川崎病(IKD),予IVIG 2 g·kg-1(分2 d)、口服阿司匹林(3.5 mg·kg-1,每天1次),并同时予拉氧头孢。后患儿体温渐正常,口唇皲裂渐好转,出现趾端及肛周蜕皮(1E),修正诊断为川崎病(KD)。住院第13 d、第18 d复查心脏彩超均未见异常,未见冠脉改变。治疗20 d后出院继续阿司匹林口服。整个病程未出现结膜充血。门诊随访3个月,复查血常规及CRP未见异常,心脏彩超未见冠脉扩张。
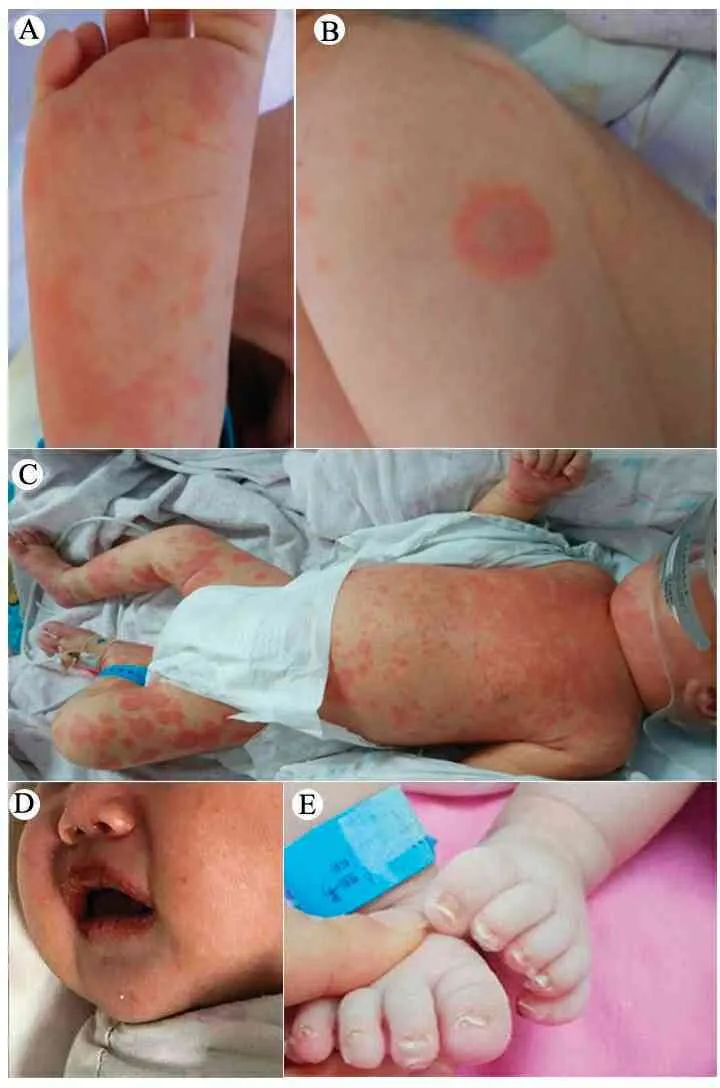
图1 入院情况
2 讨论
KD高发年龄在1岁内,<6月龄婴儿占所有KD患儿的4%~17%[1, 2]。新生儿KD罕见,即使在KD高发的日本,新生儿KD报道亦不多,Hangai等[3]统计显示,2001年1月至2012年12月日本130 243例KD患儿中仅23例为新生儿(1/5 500),在<12月的小婴儿KD中所占比例为1/1 500。23例中IKD 15例(65%),合并冠脉改变4例(17%)。
检索Cochrane Library、PubMed、CNKI、Web of Science中有关新生儿、川崎病、皮肤黏膜淋巴结综合征、neonate、Kawasaki Disease、Mucocutaneous lymph node syndrome的文献,检索日期为2000年1月1日至2019年1月1日,有详细病史资料的新生儿KD文献共15篇报告了18例[3-17]。与本文病例合并后共19例新生儿KD的临床特征总结见表1。
19例新生儿KD中,男12例、女7例,发病日龄1~26 d,完全型KD 5例(26.3%),IKD 14例。与Hangai等[3]统计比例接近(完全型KD 35%)。临床表现:发热17例(89.5%,发热≥5 d者13例),早期即四肢水肿、肿胀或蜕皮13例(68.4%),皮疹16例(84.2%),口腔黏膜改变(口唇红或草莓舌)14例(73.7%),结膜充血7例(36.8%),颈部淋巴结肿大3例(15.8%)。
KD由于缺乏特征性的实验室指标,诊断主要依赖于临床表现。新生儿因免疫系统发育不完善、临床症状不典型、发病率低,不易引起重视,诊断更为困难。Okazaki等[4]报告的1例无发热新生儿KD为早产儿,以皮疹为首发表现,伴随口唇红、结膜充血及肢端水肿,予IVIG治疗有效。另1例无热新生儿KD生后第1 d即出现冠脉扩张,无KD的其他表现,在排除其他能引起冠脉扩张的疾病后,考虑IKD导致的冠脉扩张、心包积液、PLT减少及高胆红素血症[18],回顾性诊断IKD。除了发热,最常见的临床表现是肢端改变,19例中早期出现肢端水肿13例,早期无肢端水肿、经IVIG治疗后出现蜕皮4例(例4、7、13和19)。其他临床表现出现的频率由高到低依次为皮疹、口腔黏膜改变、结膜充血及颈部淋巴结肿大。由于口腔黏膜改变及结膜充血等症状常为一过性,如在询问病史或体格检查中有疏漏,则易漏诊、误诊。颈部淋巴结肿大者仅占16%,这与我院对于6月龄婴儿KD临床表现的报道结论一致[19],也与Lee等[2]和Manlhiot等[20]的文献结论相符[2, 20]。

18例新生儿KD(有1例未报告CRP值)中CRP正常6例(33%)。本文患儿CRP在急性期正常,发热4 d后开始升高;PLT入院首次检查正常,第9 d开始升高。感染相关指标的升高更常见于年长儿,给婴儿尤其是新生儿KD的诊断增加了难度。
19例新生儿KD经超声检查,10例有冠脉改变(53%),远高于年长儿及Hangai等[3]对日本12年来新生儿KD冠脉改变的统计(20%)。患儿如有冠脉改变,结合其他体征及症状诊断KD较容易,没有冠脉改变者则容易漏诊。
新生儿发热性疾病,极易误诊为败血症、病毒感染或者免疫性疾病,如果为新生儿KD则失去了IVIG治疗的最佳时机,使机体免疫反应加重,最终发展为冠脉损害。临床工作中,遇到发热>5 d、抗生素治疗无效的新生儿,应注意观察皮肤皮疹、口唇黏膜改变、肢端变化及手足硬肿等,对可疑者及时完善心脏彩超检查。新生儿KD发病率低可能与来自母体抗体的被动免疫有关[21]。另外,新生儿KD更多表现为不完全型,可能与新生儿免疫系统发育不完善有关。
