JAK3突变相关重型联合免疫缺陷症1例报告并文献复习①
2019-08-13王颍源方盼盼张耀东康文清
王颍源 方盼盼 田 亮 张耀东 康文清
(郑州大学附属儿童医院,河南省儿童医院,郑州儿童医院,郑州450018)
严重联合免疫缺陷症(Severe combined immune deficiency disease,SCID)是一种出生后早期由于T细胞分化受阻导致致死性感染的遗传异质性疾病,而免疫缺陷均是基因突变引起的,其包括由相关基因突变所致的细胞免疫和体液免疫均受累的疾病总称,随着目前诊断技术的提高,检出率较过去明显提高[1,2]。该病患儿大多数在出生后早期发病,表现为反复、严重感染,行异基因造血干细胞移植(Hematopoietic stem cell transplantation,HSCT)进行免疫重建是其主要的治疗方法,基因治疗是目前研究的热点方向。如不及时免疫重建多数患儿将死于婴儿期。本文对我院近期收治的1例SCID患儿及其父母的血液行全外显子测序检测,并同时检索JAK3相关的重型联合免疫缺陷症的相关文献进行复习。
1 临床资料
1.1病例资料 患儿,男,2月10天。因“间断咳嗽1个月,发热半月,加重伴呼吸困难半天”为代主诉于2018年3月15日入住我院,入院前1个月患儿即有咳嗽伴发热并出现反映差,吃奶差,外院按“肺炎”予以多种抗菌药物联合应用,人免疫球蛋白支持治疗,疗效差,仍反复发热、咳嗽、并进行性加重出现呼吸困难,转入我院;个人史:患儿系第2胎第2产,足月剖宫产出生,出生体重2.9 kg;家族史:父母体健,非近亲婚配,家族中无类似患儿及遗传病史。入院查体:体温:37.1℃,脉搏:186次/min,呼吸:106次/min,血压:70/38 mmHg,全身未见明显皮疹及感染灶,全身浅表淋巴结未触及,头颅外观无异常,口腔黏膜完整,双肺呼吸音粗,可闻及大量湿啰音及痰鸣音,心音稍钝,心率快,律齐,心前各瓣膜区未闻及杂音,腹胀,肝脏右肋下3 cm,脾脏肋缘下2 cm,肠鸣音减弱,余未见异常。
1.2实验室检查及免疫分型 患儿淋巴细胞计数最低为0.24×109个/L,淋巴细胞分类计数CD3+1.48%,CD4+0.66%,CD8+0.56%,CD4/CD8 1.18,CD16+CD56+14.85%,CD19+78.87%。免疫球蛋白及补体:IgG 11.37 g/L(2~5.5 g/L),IgM<0.06 g/L(0.17~0.66 g/L),IgA<0.03 g/L(0.05~0.34 g/L),IgE 13.90 U/ml(0~60 U/ml),C3 0.98 g/L(0.78~2.1 g/L),C4 0.24 g/L(0.1~0.4 g/L)。EBV-DNA定量、CMV-DNA定量、肺炎支原体抗体、血培养、呼吸道病原检测均为阴性。骨髓涂片及细胞学检查未见明显异常。胸部CT检查提示大片状阴影及磨玻璃状改变。入我院后患儿在机械通气下,同时应用美罗培南、头孢哌酮舒巴坦、利奈唑胺针效果差,行纤支镜怀疑曲霉感染,联用卡泊芬净仍无效,患儿肺部感染仍进展,对呼吸机依赖,呼吸机参数逐渐上调,吸入氧浓度由25%上调到纯氧,氧饱和度仍不能维持,共治疗2周病情无好转,肺部影像学表现进行性加重,胸腺发育小(图1);结合体液免疫、细胞免疫功能结果,考虑免疫缺陷可能,家属放弃治疗,出院前在征得家属同意并签署基因送检知情同意书后采集患儿及其父母外周血行基因测序。对患儿JAK3基因全部编码序列进行分析发现患儿JAK3基因有1个纯合突变,两个等位基因为错义突变,其测序图见图2。患儿出院后当天死亡。
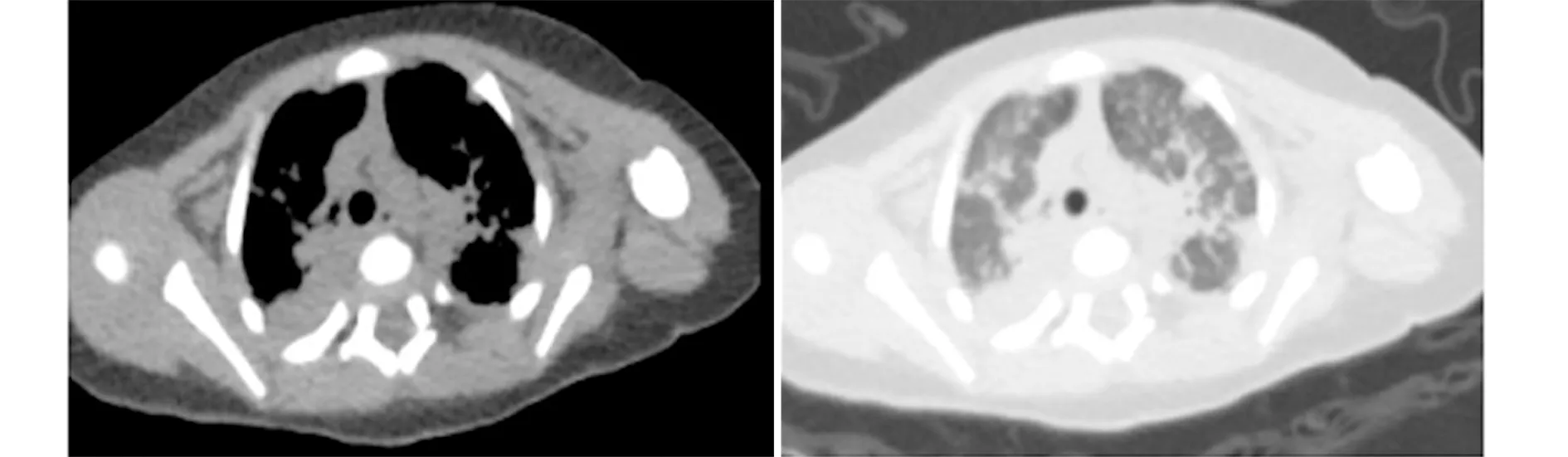
图1 患儿胸部CTFig.1 Chest CT of patient

图2 基因测序图Fig.2 Gene sequencingNote: A.The Patient;B.His father;C.His mother.
表1 JAK3相关基因突变与免疫缺陷相关报道情况
Tab.1 JAK3 related gene mutation and immunodeficiency related reports

YearCountryGene coordinateMutation informationDiseaseHgvs descriptionLiterature1995USAchr19:17948747-17948747Cys565TermSCID1695C>A[3]1995USAchr19:17951114-17951114ins 1 bp codon 393SCID1178dupG[3]1995ITAchr19:17954595-17954595Tyr100CysSCID299A>G[4]1996ITAchr19:17945378-17945378IVS16 ds T-C+2SCID2350+2T>C[5]1997ITAchr19:17949201-17949201IVS9 as A-G-2SCID1442-2A>G[6]1997ITAchr19:17947957-17947957IVS12 ds C-T-20SCID1767C>T[6]1997ITAchr19:17949199-17949199Glu481GlySCID1442A>G[6]1997ITAchr19:17950394-17950394Arg445TermSCID1333C>T[6]1997ITAchr19:17945455-17945455Cys759ArgSCID2275T>C[6]1998USAchr19:17947980-17947980Arg582TrpSCID1744C>T[7]2000ITAchr19:17942559-17942559Leu910SerSCID2729T>C[8]2000ITAchr19:17941339-17941339Tyr1023TermSCID3069C>A[8]2000ITAchr19:17941310-17941310IVS21 ds T-A+2SCID3096+2T>A[8]2000ITAchr19:17953950-17953950Pro151ArgSCID452C>G[8]2000ITAchr19:17945696-17945696Val722IleSCID2164G>A[8]2001ITAchr19:17945768-17945768Glu698TermSCID2092G>T[9]2001ITAchr19:17945434-17945434Gln766TermSCID2296C>T[9]2001ITAchr19:17943325-17943325IVS18 ds G-C+3SCID2680+3G>C[9]2001ITAchr19:17945419-17945419Arg771TermSCID2311C>T[9]2001ITAchr19:17945780-17945780Glu694LysSCID2080G>A[9]2001ITAchr19:17955226-17955226Met1ValSCID1A>G[9]2001ITAchr19:17945795-17945795Pro689SerSCID2065C>T[9]2001ITAchr19:17945988-17945988Arg651TrpSCID1951C>T[9]2001ITAchr19:17955055-17955055Ala58ProSCID172G>C[9]2001ITAchr19:17941335-17941336del 1 bp codon 1024SCID3072delC[9]2001ITAchr19:17946861-17946861IVS12 as G-A-1SCID1787-1G>A[9]2001USAchr19:17953895-17953895Asp169GluSCID507C>A[10]2001USAchr19:17955053-17955056del 3 bp codon 57SCID171_173delTGC[10]2004USAchr19:17942501-17942501Tyr929TermSCID2787T>G[11]
续表1

YearCountryGene coordinateMutation informationDiseaseHgvs descriptionLiterature2004USAchr19:17955118-17955119del 1 bp codon 36SCID108delG[11]2004USAchr19:17947959-17947959Gly589SerSCID1765G>A[11]2004USAchr19:17942054-17942054IVS20 ds C-T-18SCID2961C>T[11]2005TURchr19:17945719-17945719Thr714MetSCID2141C>T[12]2005JPNchr19:17949073-17949073Trp523TermSCID1568G>A[13]2007USAchr19:17937720-17937720IVS22 as G-A-1SCID3208-1G>A[14]2008USAchr19:17954215-17954215Pro132ThrAML related394C>A[15]2008GREchr19:17943350-17943350Tyr886TermSCID2658T>G[16]2008GREchr19:17941394-17941397del 3 bp codon 1003SCID3011_3013delTCT[16]2010CHNchr19:17955111-17955111ins 1 bp codon 39SCID115dupC[17]2010CHNchr19:17946035-17946035IVS13 as G-A-11SCID1915-11G>A[17]2013FRAchr19:17954260-17954260Arg117CysSCID349C>T[18]2013USAchr19:17953408-17953408Cys193TyrID578G>A[19]2013USAchr19:17953306-17953308del 2 bp codon 226ID678_679delCT[19]2013USAchr19:17947935-17947935IVS12 ds G-T+3ID1786+3G>T[19]2014GERchr19:17951140-17951140Ala385SerPID1153G>T[20]2015USAchr19:17946810-17946810Arg613TermSCID1837C>T[21]2016JPNchr19:17953991-17953991IVS4 as G-A-10SCID421-10G>A[22]2016JPNchr19:17953324-17953324Leu221ProSCID662T>C[23]2016BELchr19:17955112-17955112Gln39TermSCID115C>T[24]2016SAchr19:17954586-17954586Arg103HisSCID308G>A[25]2016SAchr19:17952520-17952520Gln305TermSCID913C>T[25]2016SAchr19:17951086-17951086Arg403CysSCID1207C>T[25]2016ITAchr19:17951085-17951085Arg403HisSCID1208G>A[26]2016ITAchr19:17953130-17953130Gln286TermSCID856C>T[27]2016ISRchr19:17943239-17943239IVS18 ds G-A+89SCID2680+89G>A[28]2016UKchr19:17946015-17946015Gly642ArgPID1924G>C[29]2016UKchr19:17951089-17951089Arg402CysPID1204C>T[29]2016UKchr19:17945431-17945433del 2 bp/ins 1 bp codon 766SCID2297_2298delAGinsC[29]2017TURchr19:17946779-17946779Trp623TermSCID1868G>A[30]2017TURchr19:17950343-17950343ins 1 bp codon 462SCID1383dupG[30]2017USAchr19:17943356-17943356IVS18 ds C-T-29SCID2652C>T[31]2017KORchr19:17953960-17953961del 1 bp codon 147SCID441delT[32]2017KORchr19:17953899-17953899Leu168TermSCID503T>A[32]
2 JAK3相关基因
本文检索HGMD专业数据库,目前报道的JAK3基因突变位点共63个,共涉及文献30篇,其中该基因突变是由美国和意大利率先报道,后各国逐渐报道,其中意大利报道数目最多,欧美国家占报道的绝大多数,亚洲国家报道少,我国最先报道为2010年报道2个位点,2012年报道1个位点,详细见表1。
3 讨论
严重的联合免疫缺陷(SCID)是婴幼儿一种罕见且致死的原发病,目前我国尚没有准确的SCID流行病学资料,国际上报道SCID发病率约为1/58 000[33]。免疫缺陷大多是由基因缺陷引起,如IL2RG、JAK3、IL7RA、RAG1、RAG2和ADA。目前文献报道JAK3基因突变所致的重症联合免疫缺陷症约占所有SCID的10%[34],由Macchi[3]和Russell[4]等先后报道,JAK3位于19号染色体P12-13.1,开放编码框有3 372个碱基,共有24个外显子,编码1 124 个氨基酸,以JAK3及SCID为关键词进行检索,截至2017年检索相关数据库全世界共有63个JAK3指定突变位点,且以错义突变为主,63个突变位点中大部分均由欧美国家所报道,其中意大利共报道25个突变位点,占40%,而亚洲国家报道极少,我国共报道3例,除表1中所示2例外还有2012年周雪莲等[35]报道的1例。JAK3是一种酪氨酸蛋白激酶,与多种细胞因子(如IL-2、IL-4、IL-7、IL-9、IL-15)受体共享的γ链(γc)结合影响信号传递,它使受体发生构象变化,结合相关的JAK蛋白,从而驱动酪氨酸的转磷酸化和JAK激酶区域的激活,对细胞发育与活性具有重要调节作用[36]。其中IL-7与IL-15主要调节T细胞和NK细胞的分化和活化,故JAK3缺陷致该两种细胞因子缺陷可能致T细胞和NK细胞的功能严重缺陷[37]; JAK3与JAK1、JAK2、TyK2共同组成JAK家族,JAK家族由7个结构域组成,N端的JH5、JH6与JH7又称为FERM,共编码300个氨基酸,介导部分细胞因子及其受体之间的相互作用,为JAKs与γc受体相结合组成调节蛋白的催化活性所必需[38]。JH1对调节激酶活性有重要作用,为激酶域。JH2为伪激酶结构域仅有某些调节作用[39]。故不同部位突变可导致不同程度JAK3蛋白表达以及激酶活性。相应的突变所致免疫缺陷患儿临床表现差异也较大,轻者免疫功能和临床表现可正常,只有在基因筛查时发现,重者表现为反复、严重细菌和病毒感染等典型SCID表现,多数经基因确诊的SCID的致病突变发生在JH1和JH2的结构域中[36],本例患儿入院后即表现为全身的重症感染,可疑结核感染、巨细胞病毒感染、呼吸道合胞病毒感染,且相关指标曾指向噬血细胞综合征的可能,但最终血常规提示淋巴细胞数目极少,考虑免疫缺陷的可能。
SCID 属于细胞和体液免疫严重受损的一类疾病,存在继发各种致死性感染的巨大风险,其早期识别和早期诊断显得至关重要,据报道近年在美国率先开展基于人群的新生儿SCID筛查工作,并取得良好效果[40],对疑诊SCID新生儿和小婴儿,对其进行外周血淋巴细胞亚类的流式细胞术分析和T细胞功能检测。B细胞和NK细胞数量检测结果,将有助于SCID分型和致病基因鉴定。JAK3缺陷的诊断主要通过详细询问病史及体格检查,结合性别、血常规、淋巴细胞分类计数、免疫球蛋白测定等临床诊断,最后通过JAK3基因序列分析、蛋白表达水平及STAT5磷酸化分析综合确诊。本例患儿通过全外显子检测发现其JAK3序列有1个纯合突变,c.2324G>A(编码区第2324号核苷酸由鸟嘌呤变异为腺嘌呤),导致氨基酸改变p.R775H(第775号氨基酸由精氨酸变异为组氨酸),为错义突变,该变异未在正常人群数据库中出现,经蛋白功能预测软件SIFT、PolyPhen_2、REVEL的预测结果均为致病性变异,在HGMD专业版数据库中未见报道,经家系验证分析,受检人之父该位点杂合变异,受检人之母该位点杂合变异,结合患儿临床表现及免疫学表型,我们认为上述JAK3遗传变异为其致病突变,患儿JAK3相关重型联合免疫缺陷症诊断明确。对于SCID的治疗,主要包括两个方面,非移植治疗和HSCT,非移植治疗主要包括抗感染、免疫球蛋白替代、酶替代治疗,在基因治疗尚未普遍进入临床应用前只有造血干细胞移植(Hemato poietic stem cell transplantation,HSCT)能够根治SCID[41]。文献报道如果不对SCID患儿进行相应有效的治疗,几乎100%SCID患儿将于2岁前死亡[42]。欧洲PID移植组报道已有超过1 500例SCID患者进行了HSCT[43],且其存活率逐年提高。随着技术的进步,基因治疗必将成为HSCT的一个最重要补充。
