副干酪乳杆菌的基因多样性及其抗生素耐受性分析
2019-08-12李晓姝殷瑞敏毛丙永崔树茂赵建新
李晓姝,殷瑞敏,毛丙永,崔树茂,赵建新
(江南大学 食品学院,江苏 无锡,214122)
副干酪乳杆菌(Lactobacillusparacasei)是干酪乳杆菌组群中的一个物种,与干酪乳杆菌和鼠李糖乳杆菌在糖原利用、基因序列特征等方面较为相近[1-2]。副干酪乳杆菌分布较广,主要存在于人体肠道[3]、口腔[4]、生殖道[5]等,也常出现在乳制品[6]和发酵食品[7-8]中。2010年,卫生部办公厅发布的《关于印发可用于食品的菌种名单的通知》(卫办监督发〔2010〕 65号)中包括副干酪乳杆菌。多项研究表明,副干酪乳杆菌具有抑制肠道病原菌增殖、促进肠道蠕动和提高机体免疫等益生功效[9-11],愈发成为人们研究的热点。
近年来,关于副干酪乳杆菌的菌株水平差异和遗传多样性的研究越来越广泛。STEFANOVIC等[12]发现干酪乳杆菌组群内的菌株具有遗传和代谢的多样性,SMOKVINA等[13]采用比较基因组学技术分析发现副干酪乳杆菌的进化不总是与生态位适应相关,BAO等[14]采用多位点序列分型(multilocus sequence typing,MLST)的方法分析了224株分离菌株和5株参考菌株,发现菌株遗传进化与地区或食物来源不相关。随着基因组测序技术的不断发展,采用基因组学的方法研究细菌物种多样性及遗传进化关系将成为研究重点,SUN等[15]收集了213株乳杆菌属的模式菌株,综合比较基因组学和功能基因组学的分析方法,揭示了菌株分化和功能基因随菌株进化的过程,拓展了乳杆菌的生物应用潜力。
在益生菌的安全性评价过程中,耐药性评估是菌株筛选的重要标准之一。某些具有高耐受能力的菌株可在抗生素治疗疾病时继续发挥作用,但同样也存在将抗生素抗性基因转移至病原菌的风险[16]。因此,研究副干酪乳杆菌的抗生素抗性基因的分布及菌株的抗生素耐受性规律是非常有必要的。
本研究采用基因组学的方法研究了不同地区、不同来源的33株副干酪乳杆菌的基因组特征,分析其核心基因、遗传进化关系及抗生素抗性基因,同时通过耐受性实验表征了副干酪乳杆菌的抗生素耐受能力,并与其携带的抗性基因进行关联分析,为副干酪乳杆菌的基因组学及生物学特性等研究提供了数据参考和理论基础。
1 材料与方法
1.1 菌株
33株副干酪乳杆菌,分离自中国重庆市和青海省的人或动物的粪便及泡菜样品,保藏于江南大学食品生物技术研究中心的菌种保藏中心。
1.2 主要试剂和仪器
LBS培养基、MRS培养基,购自国药集团化学试剂有限公司;IST培养基、环丙沙星、庆大霉素、新霉素、卡那霉素、氨苄青霉素、四环素、利福平、链霉素、氯霉素、红霉素、阿莫西林和克林霉素,购自生工生物工程(上海)股份有限公司。
酶标仪,美国Thermo Fisher Scientific公司;UV1800紫外可见分光光度计,日本岛津公司;Illumina HiSeq 2000测序仪,美国Illumina公司。
1.3 菌株活化及DNA的提取
将33株副干酪乳杆菌在MRS液体培养基上连续活化3代,37 ℃培养16 h,接种1 mL菌液至100 mL MRS液体培养基中培养18~24 h,取菌液在8 000 r/min条件下离心5 min并收集菌体。采用细菌基因组DNA试剂盒提取基因组DNA,操作步骤参考试剂盒说明书。DNA提取后进行质量鉴定,在浓度和纯度达到测序要求后进行测序。其中,DNA提取及质控由上海美吉生物医药科技有限公司完成。
1.4 基因组测序、组装和基因预测
使用Illumina HiSeq 2000对33株副干酪乳杆菌进行测序,得到基因组草图[17],测序部分由上海美吉生物医药科技有限公司完成。SOAPdenovo v2.0软件用于从头组装[18],用Gap Closer软件填补重叠群之间的内部空白,移除500 bp以下的读长部分,用GeneMark.HMM[19]软件预测蛋白质编码的开放阅读框(open reading frame,ORF),在COG数据库[20]和NCBI NR数据库上通过BLASTP注释ORF。
1.5 泛基因组和核心基因组曲线及韦恩图的绘制
PGAP v1.2.1[21]软件用于统计副干酪乳杆菌的泛基因组和核心基因组,所有基因被分配到核心基因组或非必需基因组集合中,然后基于核心基因和非必需基因的数量,通过Perl脚本绘制泛基因组曲线图。
1.6 系统发育进化树的构建
使用ORTHOMCL V2.0.9软件对菌株基因组进行直系同源预测,然后使用MAFFT v7.3[22]软件比对直系同源基因,提取聚类结果,通过Perl脚本绘制韦恩图。使用系统发育推论程序包(PHYLIP)(http://evolution.genetics.washington.edu/phtlip.html)[23]构建系统发育进化树,采用邻接法[24]计算副干酪乳杆菌之间的进化距离,并用于绘制系统发育树。
1.7 基因功能注释
为了获得基因组中的所有抗生素抗性基因(AR),使用已注释序列通过本地比对工具(BLAST,basic local alignment search tool)(https://blast.ncbi.nlm.nih.gov)来查询综合抗生素抗性数据库(com-prehencive antibiotic research database,CARD1.0.6,https://arpcard.mcmaster.ca)[25],若基因在CARD中的序列匹配度达到氨基酸序列同一性阈值的20%,则该基因将被认为是推定的AR决定簇。
1.8 抗生素耐受能力测定
参照欧洲抗菌药物敏感性试验委员会(European Committeeon Antimicrobial Suseeptibility Testing, EUCAST,http:∥www.eucast.org)、临床和实验室标准研究所(CLSI; www.clsi.org)和ISO标准ISO10932—2010[26],测定12种抗生素对副干酪乳杆菌的最低抑菌浓度(minimam inhibitory conceutration,MIC),抗生素包括环丙沙星、庆大霉素、新霉素、卡那霉素、氨苄青霉素、四环素、利福平、链霉素、氯霉素、红霉素、阿莫西林和克林霉素。
MIC值的测定在乳酸菌敏感性试验培养基(lactobacillus susceptibility MRS,LSM)中进行。参照ISO10932-2010 (IDF223:2010)文件,LSM培养基由IST和MRS培养基按体积比9∶1混合制成。采用二倍数稀释法,选择相应的溶剂配置不同浓度抗生素稀释液(表1);在无菌的96孔聚苯乙烯板中,每行第1孔加200 μL培养基为空白对照,第2孔~第11孔加抗生素稀释液(浓度由低到高),第12孔不加LSM培养基(不含抗生素)作为生长对照,每孔100 μL。将菌株培养至稳定期,测菌液吸光度OD625,根据OD625值预估菌液的稀释比例,使得稀释后的菌液浓度在105~106CFU/mL(OD625=1的参考浓度为3×108CFU/mL)。将稀释的菌悬液在第2孔~第12孔中加100 μL,密封后置于厌氧工作站中37 ℃培养,48 h后取出用酶标仪测定OD625。比较不同浓度下的OD625,菌株不出现生长的抗生素浓度即为该抗生素对菌株的MIC值。同一抗生素浓度,做平行3个孔;做2次独立重复试验,最终确定抗生素对菌株的MIC值。

表1 不同抗生素的稀释浓度设置Table 1 Dilution concentration setting for different antibiotics
2 结果与分析
2.1 菌株信息和基因组序列概况
本研究共有33株副干酪乳杆菌,其中12株分离自中国重庆泡菜样品,10株分离自中国重庆粪便样品,11株分离自中国青海粪便样品。在21份粪便样品中,1份来自动物粪便,10份分离自藏族人群,10份分离自汉族人群,年龄在8~84岁。所有菌株的基因组大小在2.64 ~3.18 Mb,鸟漂呤和胞嘧啶比率(Guanine & Cytosine,GC)含量为46.04%~46.51%,详见表2。

表2 33株副干酪乳杆菌的分离源信息及基因组概况Table 2 Information of isolation source and genomes of 33 Lactobacillus paracasei
2.2 泛基因组和核心基因组分析
在微生物领域,一个物种的全部基因被定义为泛基因组,包括核心基因组和非必需基因组。即使亲缘关系较为密切的菌种,其泛基因组也可能存在较大差异[27-28]。33株副干酪乳杆菌泛基因组曲线如图1所示,泛基因组的大小,在前13株菌的基因组中平均每株菌增长247个基因,余下20株菌中平均每株菌增长107个基因,最终大小为8 101个基因。泛基因组曲线呈现整体上升趋势,表明该泛基因组暂时是开放型的,原因可能是副干酪乳杆菌分布广泛,与外界各种遗传物质进行交换。
核心基因组是某个物种所有菌株保守的直系同源基因[13],与物种的生物学功能和主要表型特征相关,反映该物种的稳定性。由图1可知,副干酪乳杆菌基因组的核心基因曲线呈现迅速下滑趋势,并趋于平稳,在第33个基因组加入后稳定在1 945个基因,约占基因组的65%。王彦杰[29]发现78株粪肠球菌的核心基因组占总基因组的47.2%,相较之下,本研究中33株副干酪乳杆菌核心基因组所占比例较高,表明其基因多样性较低,可能与分离地区和来源较少有关。

图1 33株副干酪乳杆菌的泛基因组和核心基因组曲线Fig.1 Pan-genome and core genomes of 33 Lactobacillus paracasei
综上,副干酪乳杆菌的泛基因组暂时是开放型的,通过更多副干酪乳杆菌的全基因组的加入,或者更多基因组序列通过更准确的方式完全组装,副干酪乳杆菌基因组的数据集将会不断扩充,最终获得泛基因组的整体视图,其可能因为基因组多样性有限最终达到闭合,亦有可能由于进化方向多变或自身基因组多样而一直呈现为开放式。副干酪乳杆菌的核心基因组为闭合曲线,说明物种稳定性较高。
为评估33株副干酪乳杆菌的多样性,本研究基于基因组聚类结果分析其核心基因组群,并绘制韦恩图(图2),直系同源基因的个数显示在图中心,各菌株的特异性基因数量呈现在韦恩图的花瓣外侧,各菌株的总基因数标注在名称下方。33株副干酪乳杆菌中,直系同源基因数为1 945个;平均总基因数为2 988个, 其中FQHXN23L6的基因数最少(2 734个),VCQWX3_103L7的基因个数最多(3 217个);各菌株的特有基因数为10~141个。
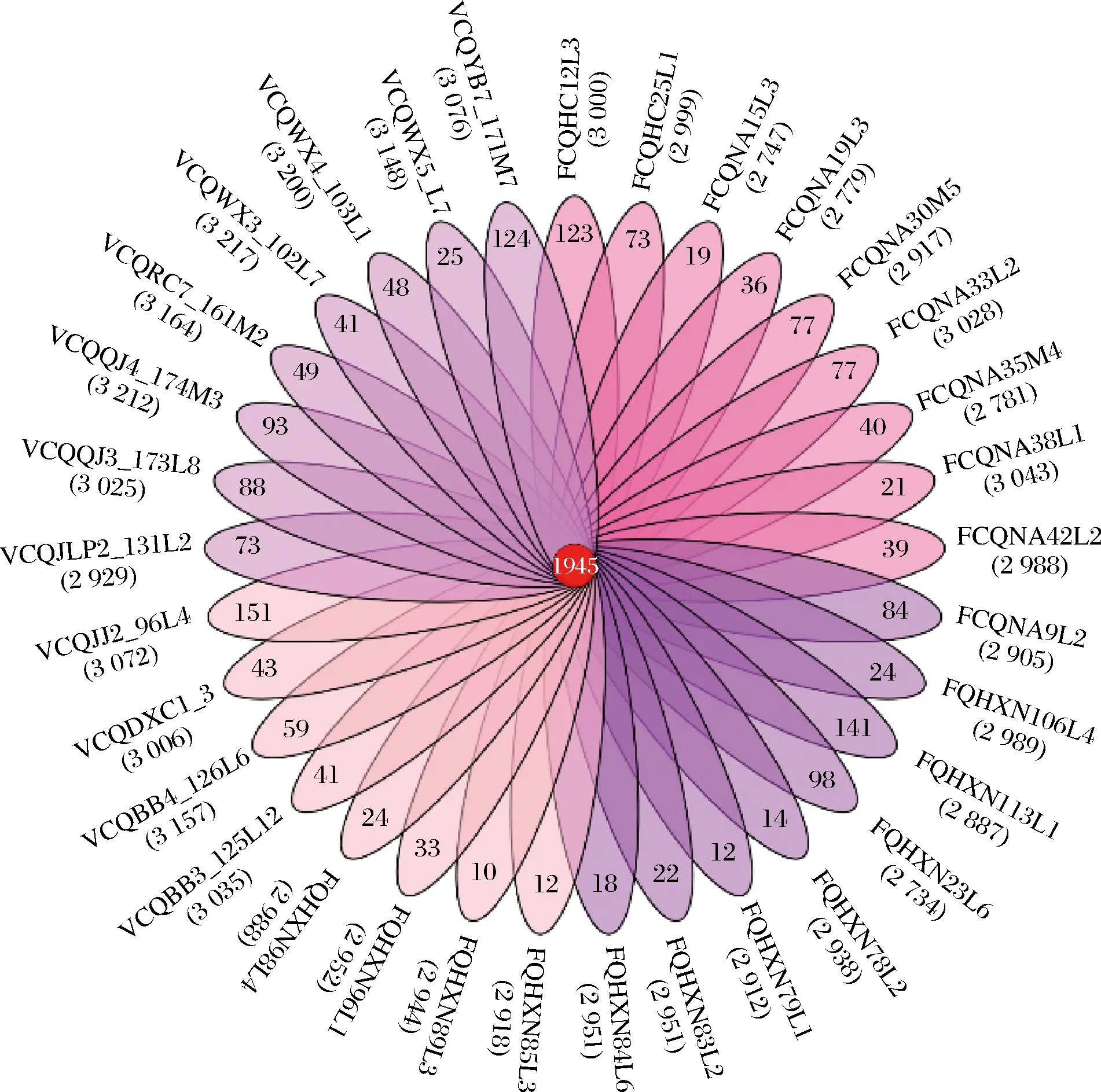
图2 33株副干酪乳杆菌的核心基因和特有基因分布Fig.2 Distribution of core genes of 33 Lactobacillus paracasei
2.3 系统发育树进化分析
为推断33株副干酪乳杆菌的亲缘关系及系统发育进化史,本研究基于直系同源基因构建了一个系统发育进化树(图3)。
图3 33株副干酪乳杆菌的系统发育进化树Fig.3 Phylogeny tree of 33 Lactobacillus paracasei
进化树被分成4个大分支,其中A分支包含11株来自青海粪便样品的菌株,其余B、C、D 3个分支包含22株来自重庆样品的菌株,表明地域差异对副干酪乳杆菌的遗传进化具有重要影响。在B、C、D3分支下,共有5个次级分支,其中2个次级分支中菌株均来自粪便样品,另外有2个次级分支中菌株均来自泡菜样品,表明不同分离宿主来源对菌株基因组进化有一定的影响。不同民族、年龄段样本的菌株在进化分支上分布规律不明显,需要增加样本数量以便探究影响副干酪乳杆菌遗传进化多样性的因素。王彦杰及BONACINA等[29-30]均提出分离源并不是影响细菌基因组进化的主要原因,这一结论与本文基本一致。综上,在副干酪乳杆菌系统发育进化方面,地域因素显示出较高影响,其次是宿主分离源。
2.4 抗生素抗性基因分析
基于耐药基因库CARD对33株副干酪乳杆菌的基因组进行比对,共发现60种抗生素抗性基因,其中有46种抗性基因存在于所有菌株中。统计各菌株中抗生素抗性基因的个数并绘制热图(图4),平均每株菌携带121个编码基因,不同菌株携带的抗性基因数没有明显差异。在分离来源、地域等因素方面,抗性基因的丰度和分布没有明显聚类,表明菌株携带抗生素抗性基因数目与来源无关。
所有菌株均携带的46种抗性基因,与红霉素、环丙沙星、氨基糖苷类、利福平等抗生素耐受相关,其中红霉素抗性基因macB丰度最高;8株分离自青海省人源粪便样本的副干酪乳杆菌不携带氯霉素抗性基因fexA;对于克林霉素抗性基因ErmB,只有FCQNA42L2和FCQNA38L1在这2株菌中存在,并且比对相似度达到98.8%;仅有FCQHC12L3这株菌携带四环素抗性基因tetM,序列相似度为90.1%。
2.5 抗生素耐受性测定
本研究测定了12种抗生素对33株菌的最小抑制浓度(MIC值),结果如图5所示。抗生素的参考断点值参照标准的EFSA指南[30]以及欧洲委员会的动物营养科学委员会(Scientific Committee on Aicmal Nutrition,SCAN)提出的建议,如果MIC值>断点值,则认为菌株对该抗生素具有抗性;如果MIC值≤断点值,则认为菌株对该抗生素敏感。
由图5可知,氨基糖苷类抗生素(新霉素、卡那霉素、庆大霉素和链霉素)的MIC值普遍高于其他8种抗生素,这与SHAO等[31]的研究结果类似。对于新霉素、利福平、卡那霉素、氨苄青霉素、氯霉素、庆大霉素和链霉素这7种抗生素,所有菌株都显示敏感结果(图5-A~图5-L),断点值分析可知,阿莫西林、环丙沙星、红霉素和四环素这4种抗生素,绝大多数菌株(32/33)都对其敏感(图5-C~图5-J),这与DANIELSEN等[32]的研究结果一致,乳杆菌属一般对能够抑制蛋白质合成的抗生素较敏感。对于克林霉素,大多数菌株(31/33)表现出敏感性(图5-B)。由此可见,副干酪乳杆菌对所研究的12种抗生素总体上都是敏感的,只有极少菌株会显示出抗性。
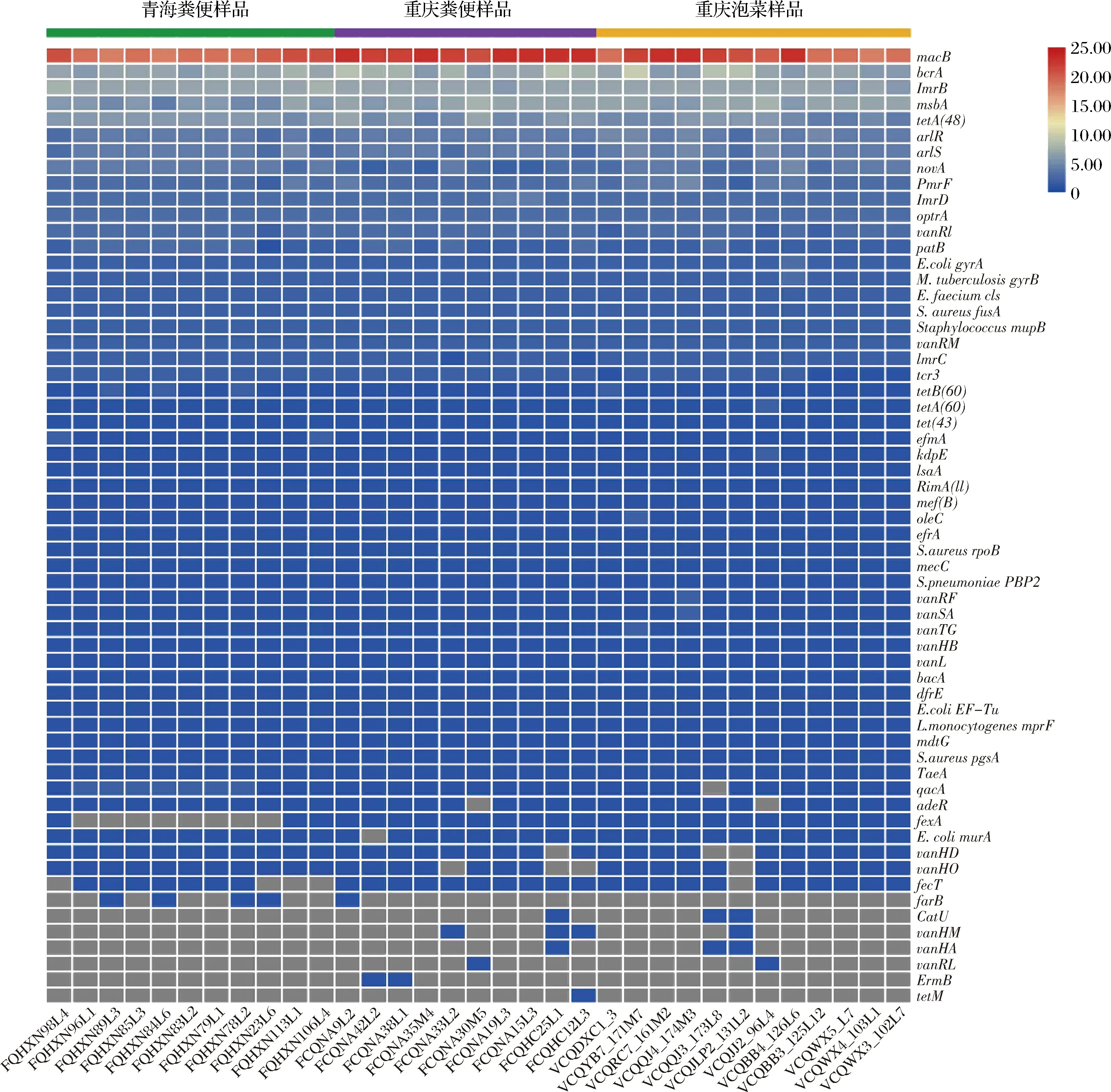
图4 33株副干酪乳杆菌携带抗性基因个数分布Fig.4 Antibiotic resistance(AR) gene counts of 33 Lactobacillus paracasei
从MIC值的分布趋势来看,除克林霉素外,其余11种抗生素的MIC值均呈单峰分布,表明不同副干酪乳杆菌对抗生素耐受浓度相对较为集中。从MIC值跨越的浓度范围来看,除氯霉素外,其余11种抗生素的MIC值跨度均较大,尤其克林霉素和四环素的MIC值范围跨越了6个浓度,表明副干酪乳杆菌对这11种抗生素的耐受性存在一定差异。
为探究菌株耐药性表型的差异与菌株分离来源之间的联系,33株副干酪乳杆菌可分为重庆泡菜组、重庆粪便组和青海粪便组。由图5可知,所有泡菜组菌株对12种抗生素都敏感,且泡菜组的MIC值范围≤粪便组MIC值。尤其是卡那霉素、环丙沙星、庆大霉素和链霉素这4种抗生素,表明泡菜来源的菌株对抗生素具有更低的耐受性和更高的敏感性。33株副干酪乳杆菌中,只有部分菌株对克林霉素、阿莫西林、环丙沙星、红霉素和四环素存在抗性,并且这些抗性菌株全部来自于粪便组。其中,只有菌株FCQNA42L2和FCQNA38L1对克林霉素表现出抗性,且为最高抗性(16 μg/mL),此表型结果与前文基因结果吻合,只有这2株菌携带序列匹配度98.8%的编码克林霉素抗性基因ErmB;只有菌株FCQHC12L3对四环素表现出抗性,且为最高抗性(64 μg/mL),其携带匹配度极高的编码四环素抗性的基因tetM。这3株菌的抗生素耐受表型与抗性基因关联分析表明,抗生素抗性基因和表型存在一定的匹配性,表明基因组学研究对于表型特性分析有不可或缺的指导意义。粪便来源的副干酪乳杆菌对特定抗生素的抗性显著高于泡菜来源的菌株,可能是抗生素的高频使用导致了人或动物样本来源的菌株对抗生素具有较高的耐受性和抗性。

A-新霉素;B-克林霉素;C-阿莫西林;D-利福平;E-卡那霉素;F-氨苄青霉素;G-环丙水星;H-红霉素;I-氨霉素;J-四环素;K-庆大霉素;L-链霉素图5 33株副干酪乳杆菌对12种抗生素的MIC分布Fig.5 Distribution of MIC values for 12 antibiotics in 33 Lactobacillus paracasei注:垂直黑线代表参考断点值。
3 结论
本研究采用比较基因组学的方法,对不同分离来源的33株副干酪乳杆菌的基因组进行分析,发现泛基因组随着更多细菌基因组的加入呈整体上升趋势,核心基因组较为稳定;分离源的地域因素对副干酪乳杆菌的遗传进化具有较为显著的影响;所有菌株均携带46种抗生素抗性基因,不同菌株携带的抗性基因数没有明显差异。
对33株副干酪乳杆菌进行抗生素的耐受性测定,发现其对12种抗生素总体上都是敏感的,但不同分离来源的菌株之间存在差异。从泡菜分离的菌株对抗生素具有低耐受性和高敏感性,部分从人粪便中分离的菌株对克林霉素、阿莫西林、环丙沙星、红霉素和四环素具有抗性。
菌株FCQNA42L2和FCQNA38L1对克林霉素、FCQHC12L3对四环素均表现出最高抗性,可能与这些菌株携带克林霉素抗性基因ErmB和四环素抗性基因tetM有关。部分抗生素耐受表型结果与抗性基因分析结果一致,表明基因组学研究对于表型特性分析有重要的指导意义。
