基于DGGE和Illumina MiSeq技术解析恩施地区米酒细菌多样性
2019-08-08向凡舒折米娜何萌廖华赵慧君郭壮
向凡舒,折米娜,何萌,廖华,赵慧君,郭壮,3*
1(湖北文理学院 食品科学技术学院,鄂西北传统发酵食品研究所,湖北 襄阳,441053)2(恩施市农业局,湖北 恩施,445000) 3(恩施市公共检验检测中心,湖北 恩施,445000)
米酒又被称为酒酿、醪糟或甜酒,是一种以糯米为主要原料,通过酒曲等多种微生物的相互作用发酵而成的发酵酒[1],盛行于湖北、海南、陕西以及广西等地。目前关于米酒的研究主要集中在理化性质分析[2]、滋味品质评价[3]、发酵工艺优化[4]和新产品研发[5]等方面,同时部分研究人员对米酒的微生物组成进行了探讨,李福荣[6]对信阳市售米酒中的微生物进行了研究,结果发现Saccharomycescerivisiae为优势菌,同时还有少量的根霉菌和细菌。焦晶凯[7]利用传统分离方法在米酒中分离出Lactobacillusplantarum,Lactobacillusrhamnosus,Enterococcussp.,Saccharomycescerevisiae以及Pichiakudriavzevii。然而关于恩施地区米酒中细菌多样性的研究报道尚少。
近年来,由于传统微生物学培养方法耗时长和工作量大,更快更便捷的分子生物学手段逐渐被人们所开发利用。变性梯度凝胶电泳(denatured gradient gel electrophoresis,DGGE)是一种用来研究各种环境中微生物群落结构演化的技术,也是一种识别和分类微生物的工具[8],广泛应用于醋[9]、白酒[10]、干酪[11]、腌菜[12]和牛肉[13]微生物群落结构的揭示。较之其他二代高通量测序技术,Illumina MiSeq平台获得的高通量序列可以降低成本并增加了每个样品的测序深度,同时具有测序量大和精确度高等特点[14],目前在水质监测[15]、植物饲料[16]、发酵食品[17]、土壤环境[18]和动物肠道[19]微生物多样性解析领域有着广泛的应用。
本研究以采集自湖北恩施土家族苗族自治州的米酒为研究对象,采用DGGE与MiSeq高通量测序相结合的手段对其微生物多样性进行了研究,同时对蕴藏的乳酸菌进行了分离鉴定。通过本研究的开展,在对恩施地区米酒中细菌进行全面解析的同时,也可为米酒的产业化生产提供一定的理论支持。
1 材料与方法
1.1 材料与仪器
米酒:采集自湖北省恩施土家族苗族自治州的农户家中,所有样品均使用农家自制米酒曲发酵而成,发酵时间3~7d。
三羟甲基氨基甲烷、乙酸、乙二胺四乙酸、丙烯酰胺、甲叉双丙烯酰胺、去离子甲酰胺、尿素、过硫酸铵、四甲基乙二胺、乙醇、冰乙酸、甲醛、AgNO3、NaOH、CaCO3,国药集团化学试剂有限公司;MRS合成培养基,青岛海博生物技术有限公司;QIAGEN DNeasy Mericon Food Kit宏基因组提取试剂盒,德国QIAGEN公司;DNA marker、PCR清洁试剂盒,京科博汇智生物科技发展有限公司;2PCR×mix,南京诺唯赞生物科技有限公司;rTaq、dNTP MIX和pMD18-T,大连宝生物技术有限公司;DGGE扩增用引物ALL-GC-V3F/ALL-V3R、正向含有7个核苷酸标签(barcode)的MiSeq测序用引物338F/806R、乳酸菌鉴定用引物27F/1495R、鉴定阳性克隆用引物M13F(-47)/M13R(-48),武汉天一辉远生物科技有限公司合成。
Veriti PCR扩增仪,美国AB公司;NanoDrop 2000微量紫外分光光度计,美国NanoDrop公司;DCodeTMSystem,美国Bio-Rad公司;DYY-12电泳仪,北京六一仪器厂;MiSeq PE300高通量测序平台,美国Illumina公司;R920机架式服务器,美国DELL公司;CT15RE冷冻离心机,日本HITACHI公司;Bio-5000 plus扫描仪,上海中晶科技有限公司;DG250厌氧工作站,英国DWS公司;ECLIPSE Ci生物显微镜,日本NIKON公司。
1.2 试验方法
1.2.1 米酒中微生物宏基因组DNA提取
参照QIAGEN DNeasy mericon Food Kit试剂盒使用说明提取10 个米酒样品中(用MJ1~MJ10表示)微生物的宏基因组DNA,使用1%的琼脂糖凝胶进行电泳,并使用NanoDrop检测其浓度。
1.2.2 PCR扩增米酒样品细菌16S rRNA基因片段
将各样品宏基因组DNA浓度做适当稀释并确定浓度一致,使用带有GC夹的正向引物ALL-GC-V3F(5’-CGC CCG GGG CGC GCC CCG GGC GGC CCG GGG GCA CCG GGG GCC TAC GGG AGG CAG CAG-3’)和反向引物ALL-V3R(5’-ATT ACC GCG GCT GCT GG-3’)对样品DNA的V3区域进行16S rRNA PCR扩增。扩增体系为25 μL(μL):10×PCR buffer 2.5,dNTP mix 2,ALL-GC-V3F 0.5,ALL-V3R 0.5,DNA模板量2,rTaq酶 0.5,ddH2O 17。反应条件为95 ℃预变性4 min,95 ℃变性30 s,55 ℃退火30 s,72 ℃ 延伸30 s,30 个循环,72 ℃完全延伸 10 min。 PCR扩增结束后,用2%的琼脂糖凝胶进行电泳检测。
1.2.3 DGGE分析
将上述检测合格PCR扩增产物作为模板进行DGGE凝胶电泳。使用8%聚丙烯酰胺凝胶,上层胶为0%变性剂,下层胶为35%~65%梯度变性剂。电泳条件:温度为60 ℃,电泳缓冲液为0.5 TAE,点样量为10 μL,先120 V,78 min,使PCR扩增产物快速通过上层胶,后80 V,13 h。
1.2.4 DGGE优势条带分析
电泳结束后对凝胶进行AgNO3染色,找出特异性条带并进行回收、扩增、清洁、连接、转化和克隆鉴定,筛选出阳性克隆子送往武汉天一辉远生物有限公司进行测序。测序结果使用Bio Edit软件将序列进行拼接,并在NCBI Blast中比对查询。
1.2.5 米酒中细菌16S rRNA PCR扩增和MiSeq高通量测序
参照沈馨等方法[20],将1.2.1中样品总DNA作为模板进行16S rRNA PCR扩增。扩增体系:5×PCR buffer 4 μL,dNTP Mix 2 μL,338F和806R各0.5 μL,rTaq酶 0.4 μL,DNA模板 10 ng,ddH2O 12.6 μL。其中引物序列为338F(5’-ACTCCTACGGGAGGCAGCA-3’)/806R(5’-GGACTACHVGGGT-3’)。扩增反应条件为:95 ℃预变性3 min;95 ℃变性30 s,55 ℃退火30 s,72 ℃延伸45 s,循环30次,72 ℃完全延伸10 min。其扩增产物用1%琼脂糖凝胶电泳检测合格后,送至上海美吉生物医药科技有限公司进行MiSeq测序。
1.2.6 序列拼接和质量控制
参照王玉荣等方法[21],对MiSeq高通量下机序列进行拼接和质量控制。首先去除不合格序列,包括碱基数<50 bp、碱基错配比率>0.2、barcode存在碱基错配和引物错配碱基数>2 bp的序列,其次去除barcode和引物序列,将剩余序列合并为一个文件以完成后续分析。
1.2.7 生物信息学分析
参照王玉荣等方法[22],使用QIIME分析平台对序列进行生物信息学分析。在将各序列校准对齐后,使用UCLUST两步法归并建立操作分类单元(operation taxonomic unit,OTU)。选出代表性序列在Greengenes数据库中进行同源性比对,确定各OTU的种属地位,同时利用Chao 1指数和Shannon指数评价微生物菌群的丰度和多样性。
1.2.8 米酒中乳酸菌的分离与纯化
使用倍比稀释涂布的方法,将各样品稀释液均匀的涂布于MRS(含质量分数为1.2%~1.5%CaCO3)固体培养基上,置于DG250厌氧工作站中(85%N2,5%CO2及10%H2),37 ℃培养48 h。挑选具有透明圈且形状大小不一的特征菌落进行划线纯化2~3次,将革兰氏染色为阳性且过氧化氢酶试验为阴性的菌落进行保存。
1.2.9 米酒中乳酸菌的鉴定
提取各纯化菌株DNA,参考张晓辉等[23]的方法使用通用引物27F(5’-AGAGTTTGATCCTGGCTCAG-3’)和1495R(5’-CTACGGCTACCTTGTTACGA-3’)进行PCR扩增。将扩增产物进行检测、清洁、连接、转化和克隆鉴定,筛选出阳性克隆子送往武汉天一辉远生物有限公司进行测序。测序结果使用Bio Edit软件将序列进行拼接,并在NCBI Blast中比对查询。
1.3 数据分析
使用Bio Edit软件和MEGA 5.0软件采用比邻法构建系统发育树,使用Origin 8.5软件对MiSeq高通量测序结果中优势门相对含量、优势属相对含量和OTU出现次数进行绘图。使用Matlab 2010b软件绘制核心OTU相对含量的热图。
2 结果与分析
2.1 米酒中细菌PCR-DGGE分析
本研究首先将10 个米酒样品中细菌16S rRNA V3片段进行PCR-DGGE分析,电泳图谱如图1所示。
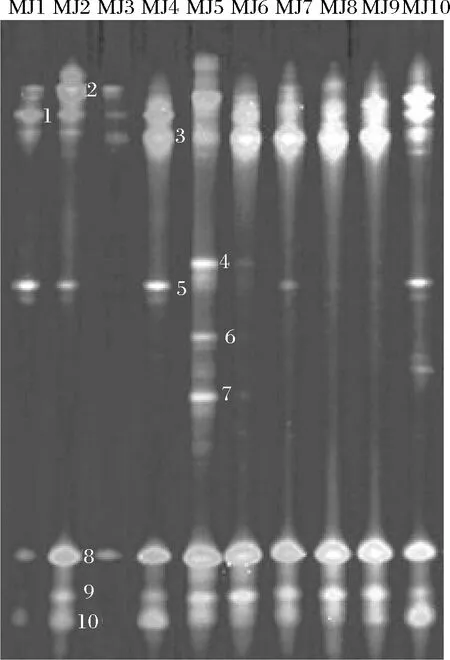
图1 米酒中细菌PCR-DGGE图谱Fig.1 PCR-DGGE analysis of bacteria in rice wine samples
由图1可知,在电泳图谱中共发现10 条特征性条带。其中条带1、2、3和8存在于每个样品中,但是亮度却不一致,说明各样品中存在相同的细菌但是丰度却不相同。条带5存在于MJ1、MJ2、MJ4、MJ7和MJ10样品中且亮度不同,条带4、6和7仅存在于MJ5样品中,条带9和10仅在MJ1和MJ3中没有表现出来。由图1可知,10 条特异性条带在MJ5样品中均有较高的亮度,说明MJ5样品中细菌群落组成具有较高多样性且各条带代表的丰度较高,而MJ3样品中仅存在4条特异性条带且亮度较暗,说明MJ3样品细菌群落组成少且丰度较低。进一步将各条带所代表的序列进行同源性比对,结果如表1所示。
由表1可知,条带1隶属于Weissella,条带2隶属于Enterococcus,条带3、8和10隶属于Pediococcus,条带4、6和7隶属于Kosakonia,同时条带5和9隶属于Lactobacillus。本试验进一步采用Neighbor-Joining(邻接法)对上述特征条带序列与数据库中16S rRNA模式菌株进行系统发育树的构建,结果如图2所示。

表1 DGGE指纹图谱中条带比对结果Table 1 The blast results of bands in DGGE fingerprint of rice wine
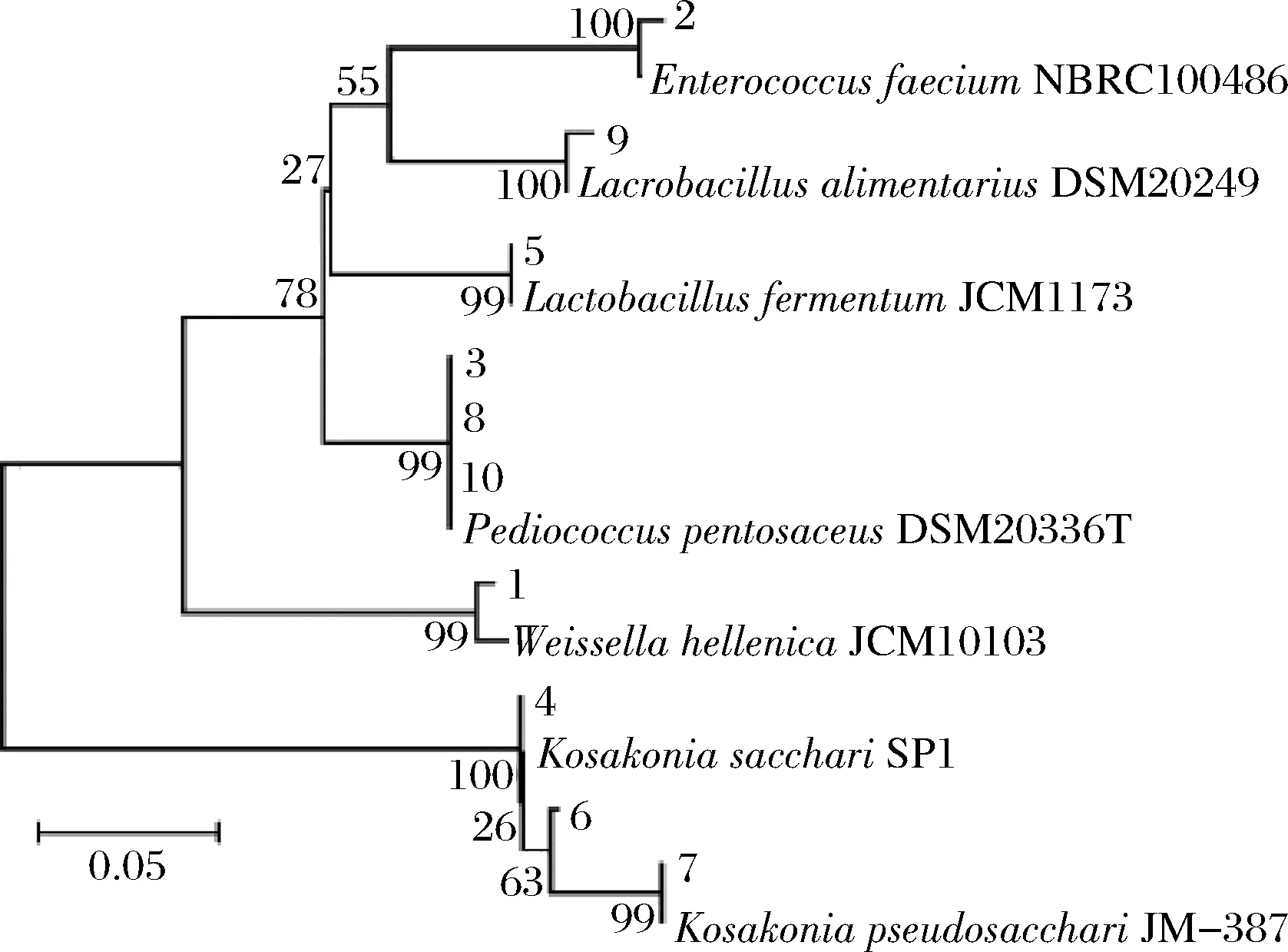
图2 DGGE指纹图谱中条带系统发育树Fig.2 Phylogenetic tree of bands in DGGE fingerprint
由图2可知,系统发育树被分为两大分支,其中条带4、6和7聚类在一个分支上,其余条带聚类在另一分支,同时发现各条带菌株代表的序列与模式菌株具有较高的相似度。值得一提的是,本研究使用引物ALL-GC-V3F/ALL-V3R,以16S rRNA的V3区为扩增靶点对细菌的多样性进行解析,然而V3区序列长度较短,为了结果准确性,本研究仅将鉴定结果明确至属水平。张振东等在16 个来源不同的米酒样品中发现Enterococcus、Streptococcus、Lactobacillus、Pediococcus以及Weissella存在于所有米酒样品中,证明了米酒样品中细菌多样性较高[24]。苗乘源等利用聚丙烯酰胺凝胶电泳(polyacrylamide gel electrophoresis,PAGE)与传统微生物培养方法相结合的手段对朝鲜传统米酒中的菌株进行分析,结果发现Lactobacillusfermentum参与米酒发酵的整个过程[25]。相飞采用PCR-DGGE技术分析发现甜酒曲中存在Weissellakimchi、Enterococcusfaecium和Herbaspirillumsp.等细菌[26]。上述3个研究的结论与本研究基本一致。
2.2 米酒中细菌MiSeq高通量测序分析
为了克服DGGE技术通量低的不足,本研究进一步使用MiSeq高通量测序技术对10 个米酒样品的细菌多样性进行了分析,其16S rRNA测序情况及各分类地位数量如表2所示。
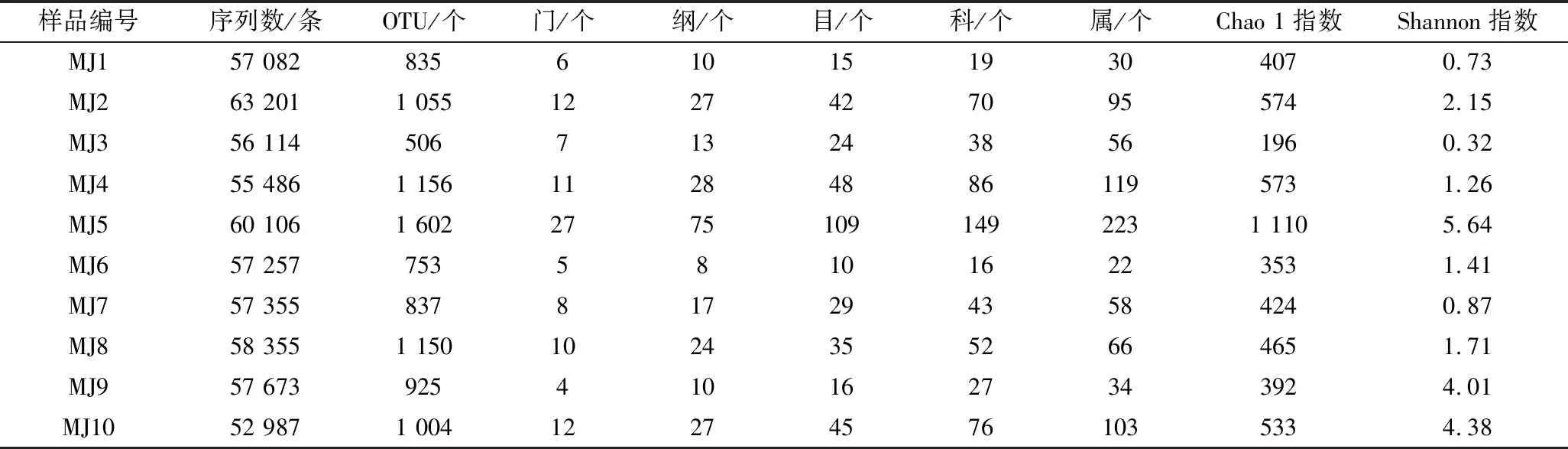
表2 样品16S rRNA测序情况及各分类地位数量Table 2 16S rRNA read counts and the number of identifiable units on different taxonomical levels
由表2可知,10 个米酒样品中共产生575 616 条序列,平均每个样品产生57 562 条序列。本试验首先根据序列的100%相似性归类获得123 817 条具有代表性的序列,根据97%相似性归类共得到7 147 个OTU,平均每个样品715 个OTU。当测序量为52 010 条序列时,MJ5样品的Chao 1指数和Shannon指数达到最大值,分别为1 110和5.64,说明此时MJ5样品中细菌群落丰富度和多样性均为最高,这与DGGE结果一致。
纳入本研究的序列被鉴定为27 个门,80 个纲,122 个目,173 个科,285 个属,仅有1.98%和8.75%未鉴定到门和属水平。米酒中平均相对含量>1%的门如图3所示。

图3 米酒中优势细菌门相对含量分析Fig.3 Analysis of relative abundance of dominant bacterial in rice wine samples at the phylum level
由图3可知,10 个米酒样品中平均相对含量>1%的门分别为Firmicutes、Proteobacteria和Bacteroidetes,其平均相对含量分别为81.53%、13.03%和2.78%。进一步分析发现Firmicutes在样品MJ6、MJ8和MJ9中较高,相对含量分别为99.84%、99.26%和99.34%,Proteobacteria在样品MJ5中较高,相对含量为87.69%。本研究进一步在属水平上对各样品的细菌多样性进行了分析,结果如图4所示。
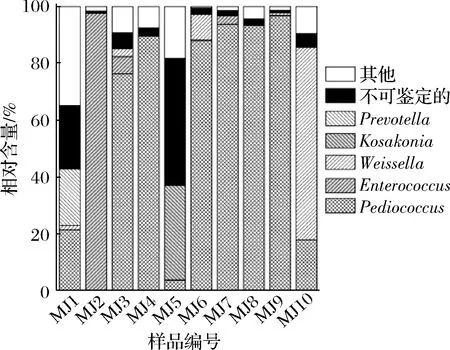
图4 米酒中优势细菌属相对含量分析Fig.4 Analysis of relative abundance of dominant bacterial in rice wine samples at the genus level
由图4可知,10 个米酒样品中共有5个细菌属的平均相对含量>1%,分别为Pediococcus、Enterococcus、Weissella、Kosakonia和Prevotella,其平均相对含量分别为58.03%、10.72%、8.24%、3.34%和2.01%。进一步研究发现,Pediococcus为样品MJ3、MJ4、MJ6、MJ7、MJ8和MJ9中含量最多的细菌,相对含量分别为76.32%、89.50%、88.02%、93.71%、93.31%和96.72%。Pediococcus和Prevotella为MJ1中的优势细菌属,相对含量分别为21.35%和19.87%;Enterococcus、Weissella和Kosakonia分别为样品MJ1、MJ10和MJ5中含量最多的细菌,相对含量分别为97.56%、67.79% 和33.32%。有研究人员亦采用高通量测序技术对米酒曲中的细菌多样性进行了解析。采用单分子实时测序技术,韩琬对3个日本米酒曲样品的细菌多样性进行了分析,结果发现米酒曲中的细菌主要隶属于Actinobacteria、Bacteroidetes、Firmicutes、Proteobacteria和Verrucomicrobia,而Proteobacteria为优势细菌门[27]。利用MiSeq高通量测序的方法,沈馨等在3个孝感凤窝酒曲中发现优势细菌属为隶属于Firmicutes的Weissella、Enterococcus、Lactococcus和Bacillus细菌属[20]。值得一提的是,MJ1和MJ5样品的细菌群落结构与其他8个样品存在较大的差异,究其原因可能与使用酒曲种类、制作环境或制作工艺有关。
本研究进一步统计了OTU在10 个米酒样品中出现次数及相对含量,结果如图5所示。
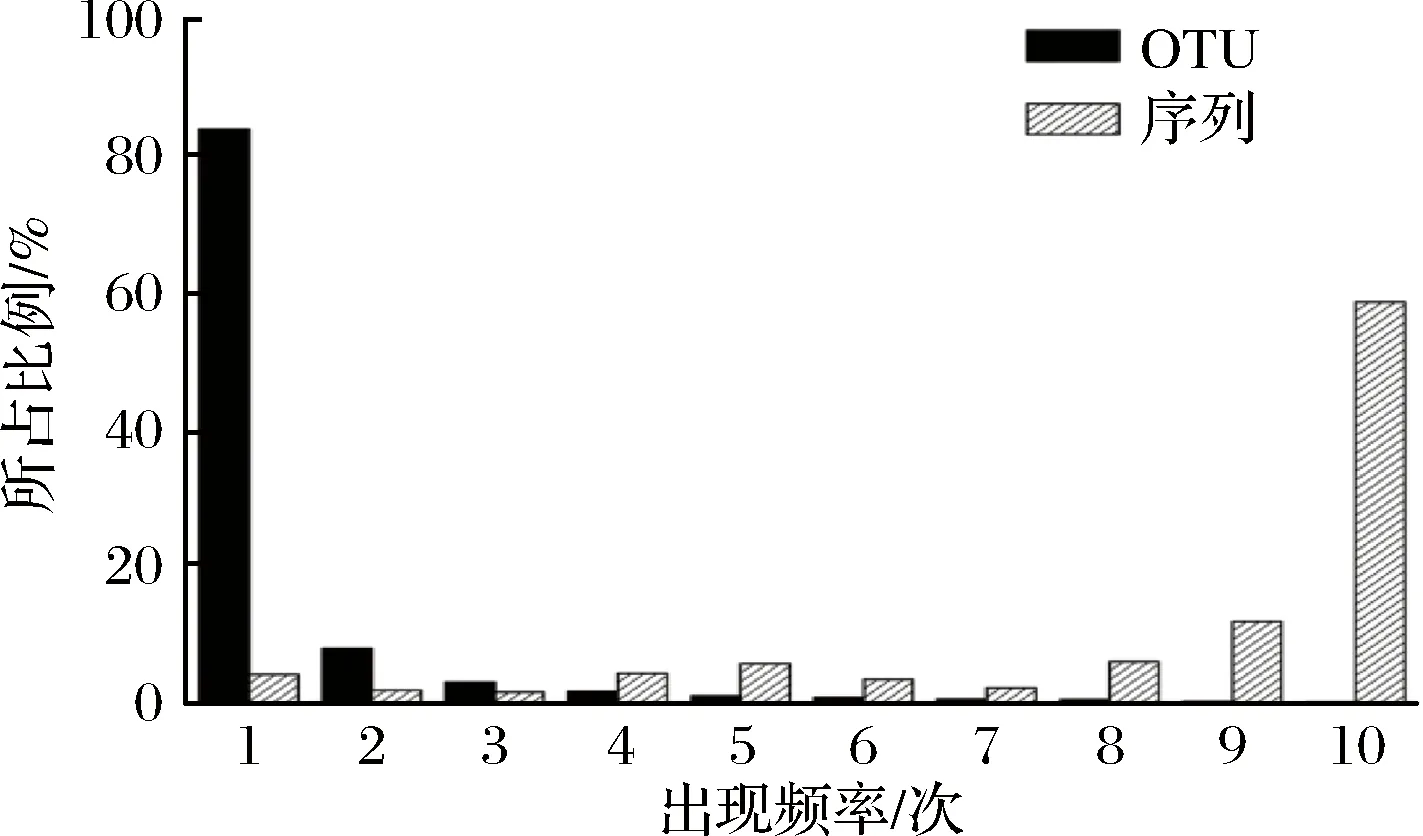
图5 OTU出现次数与其包含序列的相对含量Fig.5 Occurrence frequency and the sequence numbers included of OTU
本研究共产生7 147 个OTU,由图5可知,仅出现1次的OTU为6 008 个,占OTU总数的84.06%,但其序列仅占总序列数的4.15%。值得注意的是,出现10 次的OTU有6个,仅占OTU总数的0.08%,而序列数占总序列的58.72%。本研究进一步对核心OTU的相对含量进行了分析,结果如图6所示。

图6 核心OTU相对含量分析Fig.6 Analysis of relative abundance of core OTUs
由图6可知,6 个核心OTU分别为OTU5645,OTU59、OTU6688、OTU1966、OTU4807和OTU447,其平均相对含量分别为54.74%、0.80%、0.76%、0.33%、0.30%和0.10%。由于第二代高通量测序不能鉴定到种水平,只能确定其细菌属。进一步分析发现OTU5646隶属于Pediococcus,OTU59隶属于Ralstonia,OTU6688隶属于Herbaspirillum,OTU1966隶属于Acinetobacter,OTU4807隶属于Burkholderia和OTU447隶属于Pseudomonas。由此可见,Pediococcus为恩施地区米酒样品中的优势细菌。
2.3 米酒中乳酸菌菌株的分离与鉴定
本研究进一步对米酒中的乳酸菌进行了分离鉴定,并将测序结果与数据库中的模式菌株构建了系统发育树,结果如图7所示。
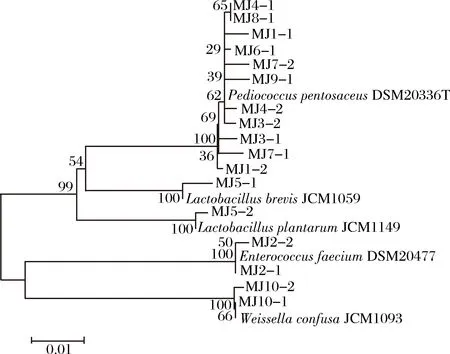
图7 米酒中乳酸菌系统发育树Fig.7 Phylogenetic tree of Lactobacillus in rice wine samples
由图7可知,在米酒中共分离鉴定出17 株乳酸菌,其中有11 株鉴定为Pediococcuspentosaceus,2 株鉴定为Enterococcusfaecium,2 株鉴定为Weissellaconfusa,1 株鉴定为Lactobacillusbrevis,1 株鉴定为Lactobacillusplantarum。由此可知,恩施地区米酒中的优势乳酸菌为Pediococcuspentosaceus,这与PCR-DGGE及MiSeq高通量测序结果一致。
3 结论
本研究以恩施地区米酒为研究对象,利用PCR-DGGE与MiSeq高通量测序技术相结合的手段对其细菌多样性进行了解析,研究发现Firmicutes、Proteobacteria和Bacteroidetes为恩施地区米酒中的优势细菌门,Pediococcus、Enterococcus、Weissella、Kosakonia和Prevotella为恩施地区米酒中的优势细菌属。此外,恩施地区米酒中乳酸菌亦具有较高的多样性,且以Pediococcuspentosaceus为主。
