基于结构域理化性质的蛋白质相互作用方向预测
2019-08-05卫博翔
卫博翔,焦 雄
(太原理工大学 生物医学工程学院,太原 030024)
蛋白质的功能必须通过其相互作用表现出来。蛋白质相互作用作为细胞生命活动中信号传递的基础,在生长、分化、代谢和凋亡中起着重要作用[1]。但是,现有蛋白质相互作用网络(PPI)通常不携带两个相互作用蛋白质之间的方向信息,如上游/下游,激活/抑制关系等。这阻碍了对生命活动中的信号传递的理解。因此,需要用相互作用蛋白质间的信号传递的方向来进一步注释当前的PPI网络。
一些计算生物学家试图从PPI网络推断信号传递方向。VINAYAGAM et al[2]基于信号通路是从质膜相关受体开始到转录因子结束的假设,从蛋白质相互作用网络中导出最短路径连接(SPC)特征,训练预测PPI网络方向的朴素贝叶斯分类器。GITTER et al[3]提出了一种优化方法来求解蛋白质相互作用网络中最大边缘方向。该方法不需要其他的信息,仅需要蛋白质相互作用网络中的拓扑信息,但是存在计算强度大的缺点。这种基于PPI网络拓扑的方法虽然简单直观,但通常没有利用KEGG和NetPath中提供的经由实验验证的蛋白质相互作用间的上游/下游信息,因此容易产生错误的信号蛋白通路和信号流方向。刘伟等[4]首先从两个相互作用的蛋白质之间的方向信息中归纳得到两个相互作用蛋白质的两个结构域之间的方向概率;然后提出了函数F来预测任何结构域对的方向,并且进一步提出了参数PIDS来预测任何相互作用蛋白质对之间信号传递的方向。但是仅凭两个结构域的方向信息尚不足以确定两个蛋白质之间的作用方向,需要考虑其他的辅助信息。梅素玉等[5]考虑到蛋白质相互作用过程中结构域的非线性组合,提取结构域特征训练一个SVM支持向量机模型,用以预测蛋白质相互作用间的激活/抑制关系。与那些基于PPI网络拓扑结构的方法相比,这些方法能够有效地利用实验验证的相互作用蛋白质之间的方向信息。
结构域作为蛋白质中具有进化保守性的结构功能单位,是蛋白质相互作用中发挥着重要作用的结构功能区域,结构域信息在方向预测中具有重要地位。本文选用结构域理化性质,提出一种基于结构域理化性质[6-7]预测蛋白质相互作用方向的新方法。该方法利用在线分析工具计算蛋白质结构域的理化性质,构成能够反映相互作用蛋白质对的特征向量,接着利用支持向量机技术分析代表这些相互作用蛋白质对的特征向量,并对其进行分类,由此预测相互作用蛋白质间信号传递的方向。本方法将蛋白质结构域自有的理化性质引入相互作用方向预测,不同于之前基于网络拓扑的方法,为以后的研究提供了一个新思路。
1 实验设计
结构域(domain)是具有一定活性的蛋白质超二级结构单元,是蛋白质折叠、设计、进化以及功能实现的基本单位[8]。一个蛋白质平均含有2~3个结构域,平均50个氨基酸构成一个蛋白质的结构域[9]。研究表明,76.4%的蛋白质具有一个或多个结构域,且结构域相互作用的方向性是广泛存在的,可以用于预测信号网络中蛋白质相互作用的方向[10]。因此,选取蛋白质结构域理化性质作为样本特征,并分析现有的具有明确方向的蛋白质相互作用数据,可以为未来进一步预测方向未知的蛋白质相互作用提供有益的帮助。
1.1 数据集
为了训练及评估分类器,首先需要用于训练和分类的标准阳性集和标准阴性集。为方便起见,本文数据集采用文献[11]中的数据集,该数据集从人、小鼠、大鼠、果蝇和酵母的所有信号网络中,分离整理出2 803对具有特定方向的蛋白质相互作用,包括激活、抑制、磷酸化、去磷酸化和泛素化,作为标准阳性集。同时,蛋白质复合物中的蛋白质相互作用被认为是不具有方向的,构建649个蛋白质复合物,将其作为标准阴性集。
1.2 构建特征向量
本文采用Expasy的protparam在线分析工具[12],得到蛋白质结构域的10种理化性质,包括氨基酸数量、分子量、理论等电点、带负电的残基总数、带正电的残基总数、消光系数、平均消光系数、不稳定指数、脂肪族指数和亲水性平均值。每一对相互作用蛋白质对的结构域均采用在pfam数据库[13]中经过注释的结构域。
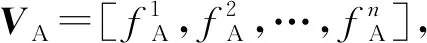
1.3 特征向量归一化
由于蛋白质结构域之间结构的差异性,计算所得的理化特性值具有一个很大的跨度,范围从几百到几万。为了避免支持向量机模型参数被分布范围较大或较小的数据支配,需要特征矩阵进行归一化处理。如公式(1)所示,特征矩阵S中的每一行对应一对相互作用蛋白质的结构域理化性质特征值,特征矩阵归一化方法如式(2)所示。
(1)
(2)
式中:Si,j表示第i对蛋白质的第j个特征值;max(S)和min(S)分别表示所有样本中第j个特征值的最大值和最小值。
1.4 基于支持向量机的蛋白质方向预测
支持向量机(support vector machine,SVM)是一种建立在结构风险最小原理基础上的机器学习方法,其可以根据样本信息在学习能力和模型的复杂性之间寻求最佳平衡。在预测蛋白质相互作用方向中,支持向量机有专门针对有限样本情况,理论上可以得到全局最优点;对于不平衡样本,能够给定一个置信水平来避免过拟合;对于高通量的蛋白质相互作用数据,能将特征向量映射到高维空间[14],计算效率高,能够进行快速的训练。
本文选用SVM分类器利用蛋白质理化性质进行蛋白质相互作用方向预测,具体步骤如下:
1) 构建特征向量。利用1.2小节的方法分别计算并构建标准阳性集和标准阴性集的相互作用蛋白质对的特征向量。最终得到的数据集样本为3 452个,其中阳性集2 803个,阴性集649个,每个样本维数为1 560维。
2) 特征向量的归一化。由于模型的输入值需在[0,1]范围内,使用1.3小节的方法对特征向量进行归一化,使特征向量的各个特征值在(0,1)范围内。
3) 生成训练集和测试集。将所有的样本集分为训练集和测试集,在标准阳性集和标准阴性集中分别随机选取4/5的数据,将这两部分组成用于训练SVM分类器模型的训练集,其余的标准阳性集数据和标准阴性集数据组成测试集。
4) 利用训练集对SVM分类器进行训练,并使用libsvm中的grid函数对模型进行参数优化。
2 实验结果与分析
2.1 评价标准
为了定量评价基于结构域理化性质预测蛋白质相互作用间信号传递方向的方法的性能,使用准确率、精确度、召回率和F-measure 4种指标来评价分类器模型的性能。4种评价指标的含义分别为:
1) 准确率(aaccuracy):正确预测的蛋白质有明确相互作用方向和无明确方向的样本数在所有样本中所占比例。
2) 精确度(bprecision):正确预测的蛋白质有明确相互作用方向的样本占所有被预测为有明确方向样本的比例。
3) 召回率(crecall):正确预测为蛋白质有明确相互作用方向占所有蛋白质相互作用有方向样本的比例。
4) F-measure(dF-measure):精确度和召回率调和均值的2倍。
4种评价指标的相关计算公式如下:
(1)
(2)
(3)
(4)
式中:PT表示预测正确的有明确方向的相互作用蛋白质对数目;NT表示预测正确的无明确方向的相互作用蛋白质对数目;PF表示将无明确方向预测为有明确方向的相互作用蛋白质对数目;NF表示将有明确方向预测为无明确方向的相互作用的蛋白质对数目。
此外,由于本文的实验数据中有明确相互作用方向的样本数据量远远大于无方向的,因此引入了接收者操作特征(receiver operating characteristic,ROC)曲线及其线下面积AUC值用于预测方法的性能评价。AUC值能更加全面地反映分类器的性能,避免由于样本数量在不同类别上的不均衡所带来的误差。
2.2 实验结果分析
SVM的核函数决定了模型的分类学习能力。目前支持向量机中常用的核函数有:线性核函数、多项式核函数、高斯径向基核函数和Sigmoid核函数。线性核函数用于线性可分的情况,具有参数少、速度快的优点,且特征空间和输入空间的维数一样,对于线性可分数据,分类效果理想;多项式核函数可以将输入空间的低维向量映射到特征空间的高维度,但是由于多项式和函数参数多,当多项式的阶数较高时,计算复杂度会阻碍分类;高斯径向基核函数局部性强,可以将一个样本映射到更高维的空间,应用范围广,对大样本或小样本都有较好的分类性能,而且相对于多项式和函数,参数较少;而采用sigmoid核函数,支持向量机实现一种多层神经网络。所以,为支持向量机模型选择恰当的核函数会达到事半功倍的效果。
首先对支持向量机的核函数进行选择,使用4/5的数据集训练分类器,剩余的1/5数据则作为测试集,测试选择不同核函数时预测模型的分类性能,实验结果如表1所示。由于实验用的数据不均衡,标准阳性集大于标准阴性集。由表1可以看出,当选择高斯径向基核函数时,分类器模型的准确率远高于其他模型,可达86.79%.因此,选择高斯径向基核函数,使支持向量机模型预测结果更加精确。
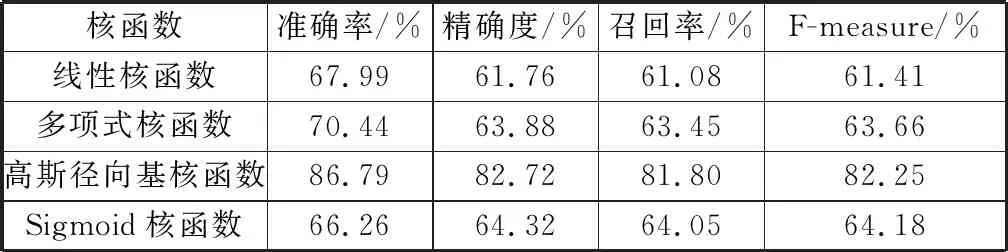
表1 不同核函数SVM预测模型的性能比较Table 1 Performance comparison of SVM prediction models with different kernel functions
进一步,使用libsvm中的grid函数来选择最佳的高斯径向基核函数的参数g和SVM的惩罚系数c.核函数参数g的网格搜索范围设置为g∈[-15,-14,…,14,15],惩罚系数c的网格搜索范围设置为c∈[-15,-14,…,14,15].如图1所示,当c=2,g=0.000 122时,该预测模型的准确率最高,为
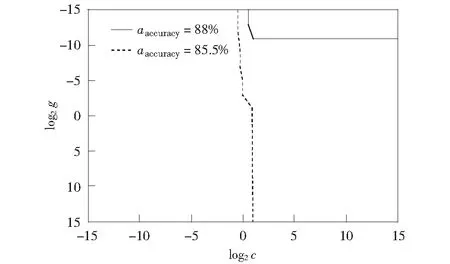
图1 c,g参数寻优结果Fig.1 c, g parameter optimization results
88.17%.因此SVM预测模型的参数选择如下:核函数选择高斯径向基核函数,高斯径向基核函数的参数g=0.000 122,惩罚系数c=2.
将上述参数用于支持向量机模型;在测试集上,该模型的预测准确率、精确度、召回率和F-measure分别为88.17%,82.94%,80.12%,81.51%.图2给出了分类器的ROC曲线,以真阳性率作为纵轴,假阳性率作为横轴,曲线下的面积越大,即AUC值越大,则分类器的性能越好。图中曲线下面积AUC值为0.837,说明分类器有很好的分类性能。这些结果表明,基于蛋白质结构域理化性质的支持向量机模型能够有效预测蛋白质相互作用间的信号传递方向。

图2 预测模型的ROC曲线Fig.2 ROC plot of the predictive model
为了进一步考察各种理化性质对蛋白质相互作用方向预测的影响,依次删除不同的理化性质,并采取5折交叉验证的方法,计算预测模型的准确率、均方误差及平方相关系数,结果见表2。由表可见,删除不同的理化性质后,预测模型的准确率、均方误差及平方相关系数均有所下降,所以蛋白质结构域的10种理化性质均有助于蛋白质相互作用方向的预测。
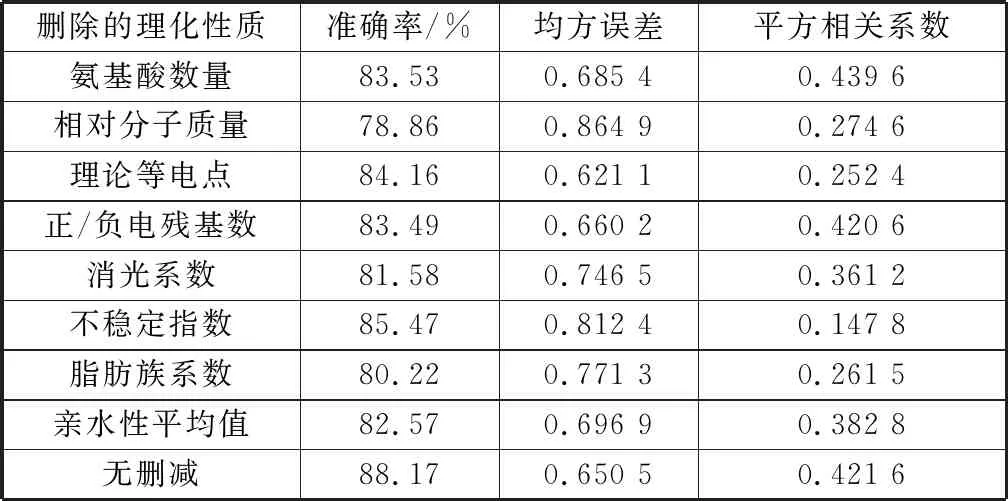
表2 删除不同理化性质后在测试集上的预测性能比较Table 2 Comparison of prediction performance on test sets after removing different physicochemical properties
为了进一步说明本文方法的可靠性,使用文献[4]中评价标准,将预测模型与PIDS方法进行对比,结果如表3所示。在准确率和误报率方面,本文的方法与PIDS方法相比略有不足;但是对于数据的覆盖度,本文的方法领先于PIDS方法。综上所述,本文提出的新方法用于预测的蛋白质相互作用间的信号传递方向是有效的。
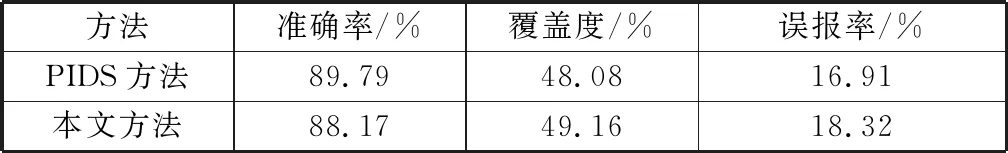
表3 不同方法的预测结果比较Table 3 Prediction results of different methods
3 结束语
笔者提出了一种基于结构域的理化特性来推断相互作用蛋白质间的信号传递方向的新方法。与以往方法相比,本文的方法关注结构域的理化性质,利用经过实验注释的具有明确方向的蛋白质相互作用信息,着重于成对相互作用蛋白质之间的信号传递方向预测。特别是,该方法可用于预测蛋白质组范围内蛋白质相互作用间的信号传递方向,并可进一步注释现有的蛋白质相互作用网络。但是此方法仍具有一定的局限性,本方法涉及到的蛋白质结构域的理化性质信息仅仅只有10种;接下来。可以尝试更多的蛋白质结构域信息,并使用特征提取方法对特征向量进行选择,来进一步完善本文提出的预测模型。
