植物样品中内源性植物激素时空分布的研究进展
2019-07-31李玉璇段春凤关亚风
李玉璇,段春凤,关亚风
(1.中国科学院分离分析化学重点实验室,中国科学院大连化学物理研究所,辽宁 大连 116023;2.中国科学院大学,北京 100049)
内源性植物激素是植物体自身合成的、控制其生长发育的一类微量或痕量信号分子。内源性植物激素是具有高度活性的小分子化合物,它们参与调控了植物体的整个生长周期,包括植物组织(或器官)的萌发、生长、分化、成熟、衰亡与休眠等过程,几乎在植物体每个生命过程中都发挥着不可替代的作用[1]。植物激素能够调节植物对外界刺激(如温度、损伤、水分等)产生应激反应,在植物体对抗外界胁迫过程中同样起到重要作用[1,2]。通过调节植物激素在植物体内的代谢过程,可以调控植物的生长发育,提高作物的品质和产量[2]。因此,植物激素的相关研究在分析化学、植物学、农业科学等领域被广泛关注。

表1 主要植物激素的理化性质及作用Table1 Physical and chemical properties and effects of the main phytohormones
pKa:acidity coefficient;logP:partition coefficient;ABA:abscisic acid;CTKs:cytokinins;GAs:gibberellins;BRs:brassinosteroids;JA:jasmonic acid;SA:salicylic acid;-:no data.
近年来我国研究者在植物激素检测方面取得了一系列研究成果,大量对植物激素的定性定量分析方法不断涌现,大大提高了我国在这一领域的技术水平[3-5]。2010年,白玉等[6]综述了5类植物激素的检测技术,并指出超微量、高灵敏的准确定性定量方法研究是未来应深入研究的一个方向;闫存玉和褚金芳团队[7]从提取纯化与分析检测方法方面综述了植物激素定量分析方法的研究进展;关亚风团队[8]对植物激素的样品前处理方法进行了综述;2017年,陈义团队[9]以超痕量植物激素检测为主线综述了当前植物激素检测方法学研究的进展。从这些综述中可以看出我国研究者在痕量植物激素分析方法研究中的贡献。
植物激素是在植物体内可迁移的有机组分,可以从植物体的产生部位运输至作用部位,进而调控植物体的生长发育。因此,植物激素在植物体内的时空分布信息是从分子层面探索植物激素调控植物生理生化作用机理的基础。如何从限量的活体植物样品中测定痕量甚至超痕量激素的时空变化,是当前内源性植物激素分析的一大挑战[8]。本文以植物激素的时空分布为主题,综述了近5年来植物激素在植物体内时空分布测定方法的相关进展,从时空分布研究的难点、分析方法和植物激素时空分布情况等几个方面进行了简要总结与讨论。
1 常见的植物激素及其分析难点
根据植物激素化学性质及其对植物体作用的不同[1],目前国际上公认将植物激素分为几大类:生长素(auxins)、脱落酸(abscisic acid,ABA)、细胞分裂素(cytokinins,CTKs)、赤霉素(gibberellins,GAs)、油菜素甾醇(brassinosteroids,BRs)、乙烯(ethylene,ETH)、水杨酸(salicylic acid,SA)、茉莉酸(jasmonic acid,JA)及其衍生物等。常见植物激素的理化性质和作用见表1,结构式见图1。

图1 主要植物激素代表物的结构式Fig.1 Structures of main representative phytohormones
不同植物激素在植物体中含量分布不同,通常在ng/g鲜重(FW)甚至pg/g FW数量级水平。生长素、ABA、CTKs、JA、SA在植物体中的含量较高[10,11],通常为1~1 000 ng/g FW;赤霉素含量为0.1~100 ng/g FW,较之含量低3~4个数量级[10];而BRs含量最低,花粉及未成熟的种子中其含量为1~100 ng/g FW,在叶片及叶芽中则低至0.01~0.1 ng/g FW[11]。植物激素通常不稳定,对环境(温度、光照、pH等)较为敏感,如赤霉素对环境pH值敏感,在偏酸性条件下才可保持较稳定状态,且当环境温度高于40 ℃时易产生变化[9,12]。植物激素多数不含可以被高灵敏检测的基团,且同类别植物激素的化学结构大多相似,存在较多同分异构现象[9]。植物体基质成分组成复杂,生物环境多变,对植物激素的分离检测造成很大干扰[8],这对植物激素的定量分析提出了更高要求。
若要获得植物激素的时空分布信息,建立分析方法时将面临以下几个难题。
(1)取样难:时空分布研究往往需要从整株植物样品中将需要分析的组织器官剥离出来。如何在剥离过程中保持样品中植物激素的原有水平和分布状态,尽可能降低甚至消除取样带来的误差,是建立时空分析方法的首要难题。
(2)称量难:与整株植物平均含量分析不同,空间分布的测定通常仅局限于植物体的某些组织器官,样品量非常有限,通常在几十微克至几毫克水平[4,10]。为获得准确的样品质量,需要高精密的天平(十万分之一甚至百万分之一天平[8])进行称量,普通的分析实验室难以实现。
(3)分析难:由于时空分布研究的样品量少,待测植物激素的质量仅在pg甚至fg数量级,这种超痕量分析对分析方法的灵敏度是一个严峻挑战。
2 植物激素的时空分布分析方法进展
2.1 取样方法
植物激素的测定通常为离体或体外分析。为了使植物样品最大限度地保持其原始激素水平,通常在采摘后用液氮将样品迅速冷冻[13-16]。液氮冷冻后的样品可以储存于-80 ℃冰箱中,待要进行样品前处理时再取出。这是目前植物激素测定的主流取样方法。而对于植物激素的时空分布测定,取样过程通常需要对植物样品的组织器官进行剥离,由此造成的损伤很可能会使植物体产生应激反应而导致植物激素水平的变化。Pan等[17]测定研究了拟南芥叶片受机械损伤后其多种植物激素的含量变化情况,发现机械损伤对JA、ABA、SA等激素的影响较大,而GAs、ZT、IAA等激素对机械损伤不敏感(见表2)。褚金芳课题组[18]对于水稻受伤叶片中6种内源性植物激素含量进行定量测定,同样发现机械损伤会对ABA、JA植物激素产生影响,而IAA、吲哚-3-丙酸(IPA)、IBA几种植物激素对机械损伤不敏感。因此,测定对机械损伤不敏感的激素时,可以在常温下对新鲜植物样品进行器官剥离或切割取样。2002年,Muller等[19]采用手术刀片在常温下对拟南芥叶片进行切割,分析其中包括IAA等酸性植物激素的分布情况。本课题组[10]此前在室温下将单片拟南芥叶片切割成15个小片,以此测定赤霉素的含量分布。而对于那些对机械损伤敏感的激素,则应采用先“固定”后取样的方案,即先采用液氮冷冻,固定激素后进行敲击剥离[4,14]植物组织器官。例如,Meulebroek等[20]在液氮环境下处理番茄叶片组织,测定其中ABA、JA、SA等8种植物激素。然而这一方法是否可以有效减少机械损伤带来的影响还有待考察。另外,对于除叶片以外的其他植物组织器官中植物激素含量受机械损伤的影响情况是否与叶片一致也需要进一步研究。
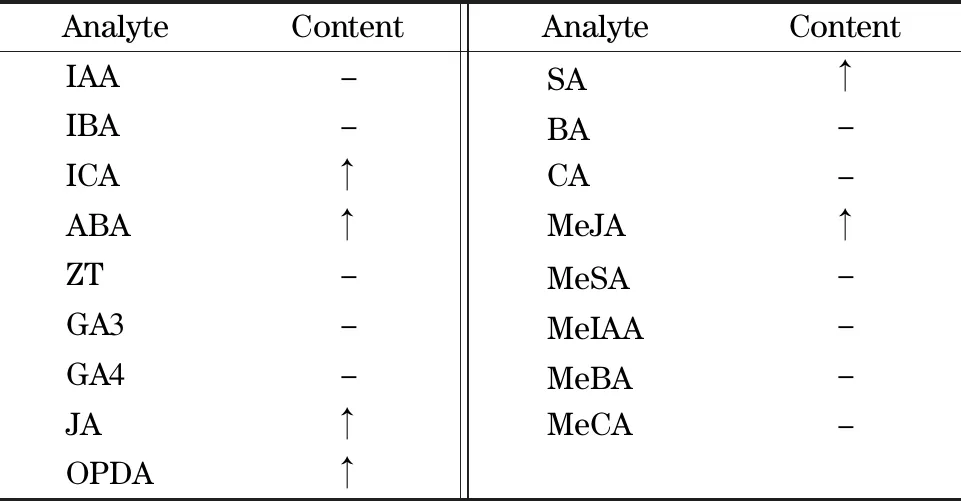
表2 损伤诱导拟南芥叶片中植物激素1 h内的变化[17]Table2 Wound-induced changes of phytohormones in Arabidopsis leaf in1 h[17]
-:no significant changes;↑:content increase;MeBA:benzoic acid methyl ester;MeCA:cinnamic acid methyl ester.
2.2 样品前处理
植物激素的时空分布测定是对微量样品的超痕量分析,存在样品量少、准确称量困难、基质干扰严重等特点,对样品前处理和仪器分析方法都提出了更高的要求。当仪器分析方法的灵敏度不足以进行单个组织器官的分析时,常常需要通过增加样本量来弥补,如从多个植株中采摘相同的组织器官合并成一个样本[21,22],这种方法测定的结果为多个植株的平均值,植株的个体差异会被掩盖,影响分析结论的准确性。近年来,国内外研究人员从提取效率、除杂方法、化学衍生等方面不断提高分析方法的灵敏度,开发出更为高效的样品前处理方法,实现了单个组织器官的植物激素测定[4,10,14]。表3汇总了近年来植物激素时空分布研究中使用的样品前处理方法。
对于微克至亚毫克样品的称重,普通的万分之一天平已经难以胜任。在缺乏更高精度天平的情况下,一般只能获得样品(某种组织器官)的平均重量,即称量多个相同的组织器官,计算单个样品重量的平均值[9]。
对于如此微量的植物样品,提高样品利用率是前处理过程的关键。目前,将植物样品在液氮环境下研磨后过夜(~12 h)浸提仍是植物激素分析中最常用的前处理步骤,通常采用甲醇、乙腈等作为萃取溶剂。然后采用液-液萃取或固相萃取等方式进行二次萃取、富集与除杂净化。例如,Xin等[26]采用95%(v/v)甲醇水溶液浸提,经SPE净化后,进行衍生后进样分析。目前固相萃取主要采用的吸附剂包括十八烷基键合相硅胶(C18)、辛基键合相硅胶(C8)、混合型强阴离子交换材料(MAX)、混合型强阳离子交换材料(MCX)和亲水-亲油平衡聚合物材料(HLB)等填料。
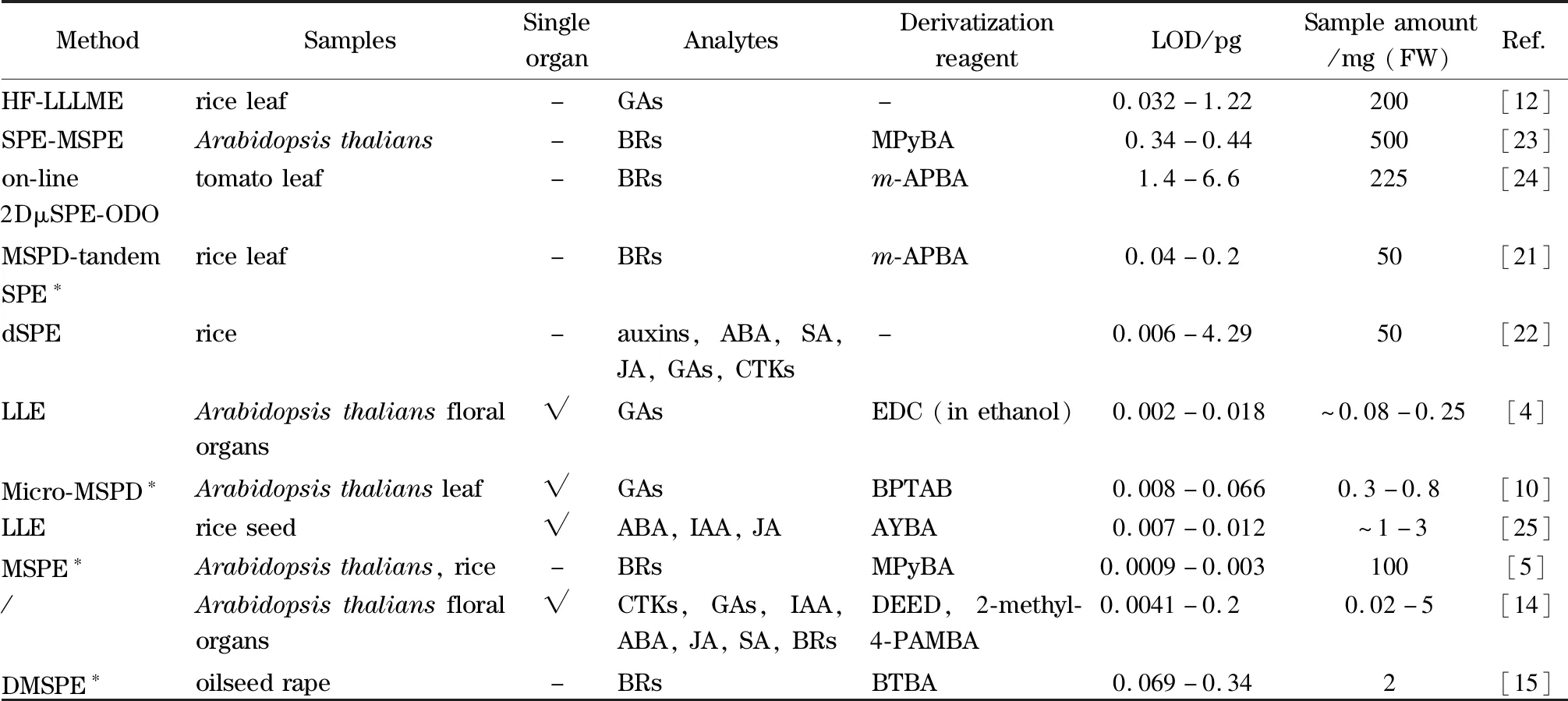
表3 近期植物激素时空分布研究中的样品前处理方法总结Table3 Summary of sample preparation method for the recent research on spatial-temporal distribution of phytohormones
FW:fresh weight;HF-LLLME:hollow fiber-liquid-liquid-liquid micro-extraction;MSPE:magnetic solid phase extraction;on-line 2DμSPE-ODO:on-line two-dimensional microscale SPE-on column derivatization;MSPD:matrix solid phase dispersion;* without overnight liquid extraction process;LLE:liquid-liquid extraction;/:without other extraction and purification steps except for overnight liquid extraction;DMSPE:dispersive matrix solid phase extraction;-:with many plant organs as the sample for determination;√:with single plant organ as the sample for determination;MPyBA:6-methoxy-3-pyridinylboronic acid;m-APBA:m-aminophenylboronic acid;EDC:N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide;BPTAB:3-bromopropyltrimethylammonium bromide;AYBA:N-(acridin-9-yl)-2-bromoacetamide;DEED:N,N-diethyl ethylenediamine;2-methyl-4-PAMBA:2-methyl-4-phenylaminomethyl-benzeneboronic acid;BTBA:4-borono-N,N,N-trimethylbenzenaminium iodide.
由于植物样品为固体,可以采用基质分散固相萃取的方式处理微量植物样品,即在液氮环境下研磨样品,而后利用SPE或分散固相萃取等方法进行净化。本课题组[21]为缩短样品前处理时间避免过夜浸提,采用了MSPD-串联SPE(MSPD-MAX-MCX)的方法实现了对水稻样品中6种BRs的测定,对比传统LLE方法,有效提高了BRs的回收率,但方法样品转移步骤较多且前处理时间较长(>1 h)。在此基础上,本课题组[10]开发了一种微型化MSPD方法,实现了在亚毫克样品中对GAs的检测。将样品置于微型离心管中于液氮环境下采用C18分散剂研磨,加入乙腈萃取后离心处理,大大减少了前处理时间(<1 h),且集提取净化于同一离心管内,处理过程中无样品转移,降低了样品损失,这对于微量样品的处理是十分有利的。而对于BRs的检测,仅仅使用C18处理无法满足对BRs测定的基质净化,故本课题组[15]在微型化MSPD的基础上加入MAX-MCX进一步净化,并发现不同复杂程度的样品基质,其最佳的分析步骤不同。这种结合了MSPD和dSPE的方法暂命名为分散基质固相萃取,它避免了传统MSPD方法中转移样品-吸附剂混合物填装SPE柱的过程,可以降低样品转移带来的损失。
为了提高微量样品中痕量植物激素检测的灵敏度,在现有的仪器条件下,采用化学衍生是目前最有效的手段。在使用MS作为检测器时,化学衍生可以有效地提高离子化效率从而提高质谱的检测灵敏度。对于常见的几类植物激素的测定,尤其是在测定植物样品中含量极低且在MS分析中低电离效率的GAs和BRs时,通常需要在进样分析前进行衍生化处理从而提高灵敏度。在时空分布的测定中,Xiao等[25]运用计算化学设计一种新型的衍生试剂N-(吖啶-9-基)-2-溴乙酰胺进一步提高羧酸类植物激素的衍生效果;Li等[4]将在乙醇环境中的N-(3-二甲基丙基)-N′-乙基碳二亚胺作为GAs的衍生试剂,实现了同时对单、双羧基的GAs的衍生,有效提高了GAs的测定灵敏度;本课题组[10]采用新型衍生试剂3-溴丙基三甲基溴化铵对GAs进行衍生,可将其MS灵敏度提高560~7 600倍,且最低可衍生水平达到1 pg/mL(~1 fg/mg),同时将BPTAB试剂与此前报道的2-溴乙基三甲基溴化铵(BETA)[27]及3-溴丙酮基三甲基溴化铵(BTA)[28]两种试剂的衍生效果进行对比,发现采用BPTAB试剂时灵敏度更高;对于BRs的衍生,本课题组[13]设计开发了一种碘化4-二羟基硼基-N,N,N-三甲基苯胺衍生试剂用于BRs化学衍生反应,将BRs的检测灵敏度提高了1 100~448 700倍;Cai等[14]采用N,N-二乙基乙二胺与2-甲基-4-苯基氨基甲基-苯硼酸两种衍生试剂用于CTKs、IAA、ABA、JA、SA、GAs及BRs的衍生化反应,实现了同时衍生处理包括BRs在内的31种植物激素。
2.3 检测方法
基于LC的高效分离能力与MS或MS/MS的高选择性和高灵敏度,LC-MS或LC-MS/MS已经成为目前植物激素分析的主流检测方法。其中MRM模式更为同时检测多种植物激素提供了便利,成为目前植物激素定量分析的首选。但是,当前植物激素标准样品的种类较少,能获得同位素标记的标准样品种类更少。为了提高现有质谱检测植物激素的可靠性,开展相关标准样品制备技术的研究是十分必要的。目前,冯钰錡团队[29,30]通过稳定同位素标记的衍生试剂衍生植物激素标准品,以其作为内标物,可以部分解决同位素内标难以获得的问题。
需要说明的是,除了本文提到的取样-前处理-色谱-质谱分离检测方案外,植物学家还通过测定与植物激素生物合成或代谢相关酶的基因表达产物,来间接获取植物激素的时空分布信息。但是这种方法很难做到同时监测多种植物激素,且通过基因表达产物无法准确获得某种激素的浓度信息。
另一方面,在植物激素的时空分布研究中成像技术也有相关的应用,如荧光成像[31]、质谱成像[32,33]等。但成像分析的局限性比较明显,比如荧光成像需要将荧光标记物引入植物体内,这个操作很可能引起植物的应激反应而产生激素水平的变化,因此反映出的浓度难以代表内源性激素的真实值。质谱成像一般只能检测植物样品表面,对于植物体内部的激素,需要切片等操作,会对植物体造成机械损伤;而且其使用的离子化手段对植物体的影响尚不清晰。因此,本文不对植物激素的成像分析展开更深入的讨论。
3 植物激素的时空分布研究
3.1 不同生长期含量变化的研究
对于植物激素在植物体内随时间变化的研究,尤其是不同生长期的植物激素的检测,可以通过采集多颗植物样品而获得较大的样品量。王泽文等[34]采用SPE与HPLC-MS/MS结合的方法测定了IAA、IBA、ABA、ZT等4种植物激素在海带薄嫩期、厚成期及成熟期的含量,发现ZT与ABA在成熟期含量最高,IAA在薄嫩期最高,而IBA含量最高出现在厚成期。Cai等[22]采用dSPE-UPLC-MS/MS方法测定了水稻在不同生长时期包括生长素、ABA、CTKs、JA、SA、GAs等54种植物激素的含量变化。Xiao等[26]采用LLE-UPLC-MS/MS监测了28 ℃温度下IAA、JA、ABA在单颗水稻种子萌发期的72 h中的含量变化,发现在种子的胚中IAA的含量在0~12 h间迅速增加,达到峰值后略有减少;ABA自萌发开始后12 h内含量急剧减少而后趋于平稳;而JA含量在萌发期缓慢增加并在60 h后趋于稳定。Li等[4]通过LLE-UPLC-MS/MS在对拟南芥花器官不同花期(花期13~15)中18种GAs含量的研究中发现,GAs含量随花期呈现出不同增减规律,如随着花期的发展沿活性GA4代谢路径中的GAs含量会有所增加。Cai等[14]采用过夜浸提结合UPLC-MS/MS定量分析了CTKs、IAA、ABA、JA、SA、GAs及BRs中12种植物激素在拟南芥花期13和15中的含量(见图2),样品量为0.02~5 mg,是目前报道的最少用量。本课题组[21]采用MSPD-串联SPE-HPLC-MS/MS测定孕穗期和成熟期水稻叶片中的BRs,发现在不同生长阶段水稻BRs具有明显的差异性,其中在孕穗期样品中扁豆甾酮(DS)含量明显高于其他几种,从孕穗期到成熟期24-表油菜素内酯的含量在增加,而DS含量明显减少。
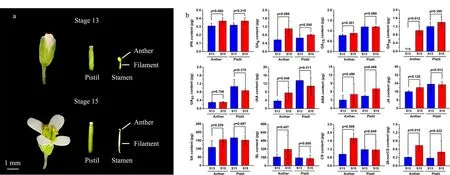
图2 花期13和15时拟南芥的(a)花器官和(b)花药、雌蕊中内源性植物激素的含量[14]Fig.2 (a)Floral organs and(b)contents of endogenous phytohormones in anther and pistil of Arabidopsis thaliana against floral stages13 and15[14]
3.2 空间及地域差异的研究
近年来在植物体的细胞、组织、器官水平上均有相关的研究工作。在植物细胞水平上,Jiskrová等[35]选取了单子叶植物拟南芥叶片和双子叶植物大麦两种模式植物的叶片细胞,采用SPE及微型SPE方法结合UPLC-MS/MS测定了CTKs除液泡外细胞内、液泡内以及细胞外间隙的空间分布,发现绝大多数CTKs分布在细胞外间隙中。在植物组织水平上,贾鹏禹等[36]通过在线SPE-HPLC-MS/MS方法测定了大豆叶片、荚皮、籽粒中IAA、ABA、ZT、GA3的含量,发现籽粒中ABA含量明显较高;张莉等[37]采用LLE-HPLC-UV方法探索了杨树枝条的尖梢、基部、上端IAA、ABA、GA、ZR等4种植物激素与枝条分枝角度发育的关系;本课题组[10]利用微型MSPD-UPLC-MS/MS方法测定拟南芥单片叶子上8种GAs的空间分布(空间分辨率为2×2 mm,样本质量0.3~0.8 mg),发现GA19的含量在单片拟南芥叶上呈现差异分布,叶柄处含量最高(见图3)。在植物的器官水平上,Li等[4]在低温下剥离拟南芥花器官(雄蕊、雌蕊、花瓣、萼片及花托),并采用LLE结合UPLC-MS/MS测定其中GAs的空间分布,发现不同植物激素在不同花器官中存在明显的差异性,如在雌蕊中GA19大量出现,疑似为GA19的合成位点;Cai等[14]采用UPLC-MS/MS检测CTKs、IAA、ABA、JA、SA、GAs及BRs的31种植物激素并定量分析其中的12种在拟南芥单花不同生殖器官中的分布;Xin等[26]采用MSPE-UPLC-MS/MS测定了BRs在拟南芥和水稻茎、叶、穗等不同器官的含量分布,探索了BRs与水稻表型的关系。
在地域差异的测定中,本课题组[15]利用DMSPE-HPLC-MS/MS测定对比了长江流域不同省份甘蓝型油菜籽中5种内源性BRs。结果表明:不同油菜素甾醇含量具有差异,其中6-脱氧-24-表油菜素甾酮含量最高且明显高于其他4种;不同省份选取的样品中,d-epiCS含量呈现差异性,其中湖北省的样品中其含量最高(见图4)。
4 结论与展望
近年来,内源性植物激素的时空分布研究取得了很大进步,但仍存在不少技术难题,对活体植物中多种植物激素的原位、实时、高分辨率检测仍未突破。目前,主流的时空分布研究仍采用活体取样-离体样品前处理-色谱-质谱分析策略,取样方式的有效性有待进一步确认;液氮研磨、长时间浸提等前处理过程限制了植物激素动态检测的时间分辨率;而空间分辨率则受取样方法、称量精度和方法灵敏度等多因素影响。另一方面,植物激素调控植物生长发育的生理机制并非单一的,而是多类激素协同作用的结果。单类激素的时空分布信息不足以反映激素调控植物生理过程的全貌。因此,当前植物激素的时空分布测定技术离植物学研究的前沿需求仍有很大差距。
笔者认为,以下几个方面可能成为该领域未来发展的主要方向。

图3 单片拟南芥叶上GAs的空间分布[10]Fig.3 Spatial distribution of GAs in an Arabidopsis thaliana leaf[10] a-c.contents of (a)GA4,(b)GA8 and (c)GA19 in different leaf disc (position was marked in Fig.3d)of a Arabidopsis thaliana leaf;d.spatial distribution of GA19 within a single Arabidopsis thaliana leaf.

图4 长江流域不同省份甘蓝型油菜籽中5种内源性油菜素甾醇的地域分布[15]Fig.4 Geographical distribution of five endogenous BRs of Brassica napus L.oilseed rape originated from different provinces in Yangtze River basin[15]
多种类植物激素的时空分布研究:目前大多数时空分布研究尤其是空间分布研究仍集中在单一种类的植物激素,为获得更全面的激素“地图”信息,有必要开发同时测定多种类植物激素的分析方法,实现多种类植物激素的时空分布测定。
无损、原位植物激素分析方法:在活体植物上进行无损、原位植物激素分析可以避免取样、样品前处理带来的不确定因素,获得最直接的激素含量和分布数据。目前,任何一种成像技术都无法完全满足植物激素时空分布研究的需求,质谱成像、光学成像具有良好的发展前景;一些原位、活体前处理方法也具有很大的发展潜力。
低等植物中植物激素的时空分布研究:伴随着植物激素测定方法的不断发展,植物学家对植物激素生理作用的关注已从陆生高等植物逐渐扩展到低等植物,如微藻[38]等;目前,对低等植物中植物激素的研究报道远少于高等植物,低等植物中多种类激素的协同作用及时空分布情况才刚起步,植物激素在低等植物与高等植物中的含量与作用差异有助于植物进化学的研究。
总而言之,原位、实时、高分辨、动态监测植物体内的痕量植物激素仍是非常具有挑战性的研究课题,也给分析科学的发展提供了较多机会。
