Comparing nanoflow reversed-phase liquid chromatography-tandem mass spectrometry and capillary zone electrophoresis-tandem mass spectrometry for top-down proteomics
2019-07-30ElijahMCCOOLLiangliangSUN
Elijah N.MCCOOL,Liangliang SUN
(Department of Chemistry,Michigan State University,East Lansing 48824,United States)
Abstract:One of the major shortcomings in top-down proteomics is the lack of efficient separations for intact proteins that can be effectively coupled to mass spectrometry.Capillary zone electrophoresis (CZE)and nanoflow reversed-phase liquid chromatography (nanoRPLC)are two methods that can be coupled to mass spectrometry directly and have been recently advanced in terms of their ability to separate intact proteins in complex biological mixtures.In this work,for the first time,we compared the state-of-the-art nanoRPLC-MS/MS and CZE-MS/MS platforms for top-down characterization of a standard protein mixture and an Escherichia coli (E.coli)proteome sample.CZE-MS produced comparable signals of standard proteins to RPLC-MS with 10-times less sample consumption.Interestingly,the proteins in RPLC-MS tended to have higher charge states than in CZE-MS,most likely due to the high acetonitrile concentration in RPLC mobile phase,leading to the more extensive unfolding of proteins in RPLC compared to in CZE.CZE-MS/MS identified 159 proteins and 513 proteoforms using 1-μg E.coli proteins in a single run and outperformed RPLC-MS/MS using 1-μg E.coli proteins in terms of protein and proteoform identifications (159 vs.105 proteins and 513 vs.277 proteoforms).The RPLC-MS/MS using 8-μg E.coli proteins identified 245 proteins and 1 004 proteoforms in a single run,and the data was much better than that from CZE-MS/MS (1-μg E.coli proteins)regarding the number of identifications because of the 8-times higher sample loading amount and significantly wider separation window of RPLC-MS/MS compared to CZE-MS/MS.
Key words:top-down proteomics;capillary zone electrophoresis- tandem mass spectrometry (CZE-MS/MS);reversed-phase liquid chromatography-tandem mass spectrometry (RPLC-MS/MS);Escherichia coli;high sensitivity
Efficient separations coupled to mass spectrometry (MS)are a vital aspect to any proteomics study characterizing complex biological mixtures [1,2].Highly efficient and high capacity separations are especially difficult to achieve when characterizing intact proteoforms,as in top-down proteomics studies [3-8].A large variety of separations have been utilized in top-down proteomics studies with trade-offs between separation efficiency,protein solubility,and compatibility with MS.The most common methods for separating intact proteins include separations based on hydrophobicity such as reversed-phase liquid chromatography (RPLC),charge-based separations including capillary zone electrophoresis (CZE)and isoelectric focusing (IEF),and size-based separations including size exclusion chromatography (SEC),gel-electrophoresis,and gel-eluted liquid fraction entrapment electrophoresis (GELFrEE)[4,5,9-12].
Advancements in RPLC have recently been made with the use of shorter bonded stationary phases (C4 and shorter),a variety of particle types and column packing procedures,the use of monoliths,and longer columns [5,13,14].The main advantages of RPLC over CZE is the loading capacity,which is consistently in the microgram range,and control over the separation window which is the window of time in which proteoforms are eluted out of the column [11,15].This is an important point when considering the large dynamic range of protein concentrations in the proteome,which can approach 7 orders of magnitude [16,17].Also,it is estimated that ~1 million unique proteoforms exist in the human proteome [18].Shen et al.demonstrated that high peak capacity (Pc~400)was possible for intact protein separation with an optimized long-column RPLC system,which,when coupled to MS,resulted in the identification of ~900 proteoforms from anS.oneidensislysate in a single run [5].Specifically,long columns (120 cm long × 100 μm i.d.)were used with superficially porous particles (3.6 μm,20 nm pores)in that study.The loading amount was 2.5 μg,and the separation window approached 800 minutes while still retaining an efficient separation.
Without the use of particles and with a complementary separation mechanism,CZE can offer additional insight into complex proteomes [19-22].The lack of particles is especially important for the separation of intact proteins to limit zone broadening and sample loss [23].Also,with current interfaces,CZE outperforms LC-MS/MS platforms in terms of sensitivity [8].An improved CZE-MS platform for the separation and identification of proteoforms has resulted in ~600 proteoform and 200 protein identifications in a single run of anEscherichiacoli(E.coli)sample [11].This CZE-MS platform,utilizing dynamic pH junction-based sample stacking,increased the loading capacity to ~1 μg and the separation window to 90 minutes [11,15,24-26].Combining offline SEC-RPLC fractionation to this optimized CZE-MS platform resulted in the identification of 5 705 proteoforms and 850 proteins from anE.colilysate [27].Fractionating before CZE-MS analysis alleviates the inherent limitations of CZE and using RPLC combines the complementary nature of these two techniques to reach unrivaled proteome coverage.Li et al.showed that,on the protein level,the overlap was less than 35% using a C18 column (100 mm long × 100 μm i.d.,1.7 μm particles)for RPLC separation and an uncoated capillary (30 cm in length)for CZE separation under high voltage (5.5 kV)[19].
Herein,we offered the first direct comparisons of RPLC-MS and CZE-MS for top-down MS characterization of a standard protein mixture and anE.coliproteome sample.
1 Experimental
1.1 Materials and reagents
MS-grade water,acetonitrile (ACN),methanol (MeOH),formic acid (FA),isopropyl alcohol (IPA)and HPLC-grade acetic acid (AA)were purchased from Fisher Scientific (Pittsburgh,PA).Ammonium bicarbonate (NH4HCO3),urea,ammonium persulfate,3-(trimethoxysilyl)propyl methacrylate,dithiothreitol (DTT),and iodoacetamide (IAA)were purchased from Sigma-Aldrich (St.Louis,MO).Hydrofluoric acid (HF,48%-51% solution in water)and acrylamide were purchased from Acros Organics (NJ,USA).Fused silica capillaries (50 μm i.d./360 μm o.d.)were purchased from Polymicro Technologies (Phoenix,AZ).Complete,mini protease inhibitor cocktail and PhosSTOP (EASYpacks)was from Roche (Indianapolis,IN).Capillary columns for RPLC were bought from CoAnn Technologies,LLC (Richland,WA).
1.2 Sample preparation
The stock standard protein mixture consisted of ubiquitin (~8.5 kDa,0.1 mg/mL),cytochrome c (cyto.c,~12 kDa,0.1 mg/mL),bovine serum albumin (BSA,~66 kDa,1 mg/mL),myoglobin (myo,~17 kDa,0.2 mg/mL),carbonic anhydrase (CA,~29 kDa,0.2 mg/mL),andβ-casein (~24 kDa,0.4 mg/mL).Mixtures were dissolved in either mobile phase (MP)A,or ammonium bicarbonate (ABC,50 mmol/L)for RPLC or CZE,respectively.The stock mixture was diluted by a factor of 10 with MP A or ABC (50 mmol/L)before analysis.
E.colisamples were prepared the same way for RPLC and CZE except for the final solvent for analysis.E.coli(K-12 MG1655)was cultured in Lysogeny broth (LB)medium (37 ℃)while shaking (225 r/min)until the OD600 reached 0.7.E.colicells were harvested through centrifugation (4 000 r/min for 10 min)and washed three times with phosphate-buffered saline (PBS).Lysis was performed in a buffer contained 8 mol/L urea,a phosphatase inhibitor,and a protease inhibitor cocktail with the assistance of sonication on ice for 15 min with a Branson Sonifier 250 from VWR Scientific (Batavia,IL)after homogenization with a Homogenizer 150 from Fisher Scientific (Pittsburgh,PA).Following centrifugation (18 000× g for 20 min),the supernatant containing extracted proteins was collected.A bicinchoninic acid (BCA)assay was performed to determine the protein concentration (~13 mg/mL).TheE.colisample was desalted using a C4 trap column (Bio-C4,3 μm,30 nm,4.0 mm i.d.,10 mm long)from Sepax Technologies,Inc.(Newark,DE)on a 1260 Infinity II HPLC system from Agilent (Santa Clara,CA).The eluate containing theE.coliproteins was collected and lyophilized.TheE.coliprotein sample was redissolved in either MP A for RPLC-ESI-MS or in an ABC buffer (50 mmol/L,pH 8)for CZE-ESI-MS.The final protein concentration was ~2 mg/mL.
1.3 RPLC-ESI
NanoRPLC was performed with an analytical column (C2,90 cm long × 100 μm i.d.,3 μm beads,30 nm pores)connected to an EASY nanoLC-1200 (Thermo Fisher Scientific).The RPLC system was connected directly to an ESI emitter with a metal union.A Spellman CZE1000R (Hauppage,NY)power supply was used to provide voltage for electrospray ionization (ESI)through the metal union.A 240-min linear gradient from 100% MP A (10% ACN,0.1% FA)to 75% MP B (70% ACN,30% IPA,0.1% FA)was used with a flow rate of 300 nL/min.The sample was loaded onto the column in MP A under a pressure of 800 bar.Blanks were performed between runs using a 120-min linear gradient (10% B-90% B)at 300 nL/min.Loading volumes were 0.5 μL for the 1 μgE.colirun and 4 μL for the 8 μgE.colirun.
1.4 CZE-ESI
An ECE-001 CE autosampler and a commercialized electro-kinetically pumped sheath flow CE-MS interface from CMP Scientific (Brooklyn,NY)was used [28-30].A fused silica capillary (1-m-long,50 μm i.d.,360 μm o.d.)was coated with linear polyacrylamide (LPA)according to references [31] and [32].The outer diameter of one end of the capillary was reduced to ~70-80 μm by etching with hydrofluoric acid based on reference [28].(Caution:use appropriate safety procedures while handling hydrofluoric acid solutions.)
Samples were injected into the capillary by applying pressure for a specified amount of time to achieve the necessary volume and sample amount based on Poiseuille’s law.Loading volume was 200 nL for the 40-ng standard protein analysis and 500 nL for the 1 μgE.coliprotein analysis.A separation voltage of 30 kV and 20 kV was used for the standard protein sample andE.colisample,respectively.The capillary was flushed between runs with background electrolyte (BGE)with a pressure of 10 psi for 10 min.A glass capillary (1.0-mm o.d.,0.75-mm i.d.,10 cm long)was pulled with a Sutter P-1000 flaming/brown micropipette puller and used as the ESI emitter.Emitter orifice was 20-40 μm with a voltage for ESI of about 2 kV.BGE for CZE was 5% (v/v)acetic acid (pH 2.4)and the sheath buffer consisted of 0.2% (v/v)formic acid and 10% (v/v)methanol.
1.5 MS and MS/MS
A Q-Exactive HF mass spectrometer was used for the experiments.The “intact protein mode” was turned on and a trapping pressure of 0.2 was used.The same MS and MS/MS settings were used for the RPLC and CZE experiments.A data dependent acquisition (DDA)method was used.Ion transfer capillary temperature was set to 320 ℃ and the s-lens RF level was 55.For full MS,the number of microscans was 3,resolution was 120 000 (atm/z200),AGC target value was 1×106,maximum injection time was 100 ms and the scan range wasm/z600-2 000.For MS/MS,the number of microscans was 3,resolution was 120 000 (atm/z200),AGC target value was 1×105,and maximum injection time was 200 ms.The top 5 most intense ions,for data dependent acquisition (Top 5 DDA),in full MS spectra were isolated with a 4m/zwindow and sequentially fragmented at normalized collision energy (NCE)of 20%.The intensity threshold for triggering fragmentation was 1×105.Charge exclusion and exclude isotopes settings were turned on with proteins with charge state higher than 5 able to be fragmented.Dynamic exclusion was used with a setting of 30 s.
1.6 Data analysis
For standard protein intensity analysis,base peak electropherograms with 5-point Gaussian smoothing were used.ForE.colisample data analysis,TopFD (TOP-down MS feature detection)and TopPIC (TOP-down MS based proteoform identification and characterization)were used for database searching [33].Raw files were converted into mzML files with the Msconvert tool followed by spectral deconvolution performed with TopFD to generate msalign files for database search using TopPIC (version 1.2.3)[34].TheE.coli(strain K12)UniProt database (UP000000625,4 313 entries,version June 28,2018)was used for database searching.Maximum number of unexpected modifications was 2,precursor and fragment mass error tolerances were 15 ppm (15×10-6),and the maximum mass shift of unknown modifications was 500 Da.The false discovery rates (FDRs)were estimated using the target-decoy approach [35,36].The proteoform identifications were filtered with a 5% proteoform-level FDR.
2 Results and discussion
2.1 RPLC-MS and CZE-MS for analysis of a standard protein mixture

Fig.1 Standard protein mixture data from CZE-MS and RPLC-MS a.Base peak electropherogram of the protein mixture using CZE-MS.40 ng of proteins was injected for analysis.b.Base peak chromatogram of the protein mixture using RPLC-MS.400 ng of proteins was injected for analysis.c.Charge state distributions of myo,CA,and β-casein using CZE-MS.d.Charge state distributions of myo,CA,and β-casein using RPLC-MS.
We first employed a standard protein mixture containing proteins with molecular weight in a range of 8.5-66 kDa for the comparison of RPLC-MS and CZE-MS in terms of separation,signal intensity,and charge state distributions of proteins.For CZE-MS,we employed an LPA-coated capillary to reduce the protein adsorption on the inner wall of the capillary and to reach a wider separation window [11].We used a dynamic pH junction-based sample stacking method for online concentration of the proteins based on our recent work [11].For RPLC-MS,we employed a 90-cm-long nanoRPLC column (100-μm i.d.)packed with 3 μm C2 porous beads (30 nm pores).We got the nanoRPLC column from Dr.Yufeng Shen at CoAnn Technologies,LLC (Richland,WA).Based on reference [5],the column represents one of the state-of-the-art nanoRPLC columns for the separation of complex intact protein mixtures.We employed a Q-Exactive HF mass spectrometer in the experiment.The MS parameters for CZE-MS and RPLC-MS were the same.
Fig.1 shows the separation profiles and charge state distributions of proteins using CZE-MS and RPLC-MS.First,CZE and RPLC have different mechanisms for protein separation,size-to-charge ratio vs.hydrophobicity,leading to drastically different migration or elution orders,as shown in Figs.1a and 1b.For example,BSA migrated fastest in CZE and had stronger retention than ubiquitin and cyto.c in RPLC.It suggests that a combination of RPLC and CZE can produce orthogonal and high capacity separation of complex mixtures of intact proteins.We recently demonstrated the power of nanoRPLC-CZE-MS/MS for orthogonal and high capacity separation of an MCF7 cancer cell proteome digest,leading to the identifications of nearly 8 000 proteins and 60 000 peptides starting from only 5-μg peptides [37].We noted that RPLC-MS produced a very broad peak of cyto.c (over 10-min wide),and CZE-MS yielded a reasonably sharp peak of cyto.c (less than 1-min wide).The broad peak of cyto.c in RPLC-MS might be due to its weak retention on the C2 column,leading to its inefficient trapping at the front end of the column during sample loading.Second,CZE-MS had much higher sensitivity than RPLC-MS.CZE-MS with 40-ng proteins injected produced comparable protein intensity and signal-to-noise ratio to RPLC-MS with 400-ng proteins injected,as shown in Figs.1a,1b,and Table 1.The data agreed well with that from Yates group recently [8].The high sensitivity of CZE-MS makes it extremely useful for top-down proteomics of mass-limited samples.Very recently,we showed that thousands of proteoforms were identified from zebrafish brains using advanced CZE-MS/MS with only 500-ng protein material [38].Third,interestingly,the proteins in RPLC-MS tended to have higher charge states than that in CZE-MS,as shown in Figs.1c and 1d.For example,the most abundant charge states of CA in CZE-MS and RPLC-MS are +31 and +35,respectively.This phenomenon is most likely due to the high acetonitrile concentration in RPLC mobile phase,leading to the more extensive unfolding of proteins in RPLC compared to that in CZE.Higher charge states can potentially benefit gas-phase fragmentation of proteins for identifications due to their more unfolded structures.Fourth,RPLC-MS produced a much wider separation window than CZE-MS for the standard protein mixture,80 min vs.20 min,as shown in Figs.1a and 1b.Because we employed a 90-cm-long RPLC column,we used a 240-min gradient for separation,leading to a wide separation window.This feature becomes one very important advantage of RPLC-MS compared to CZE-MS for top-down proteomics.For large-scale top-down proteomics,a wide separation window is vital because more MS/MS spectra can be acquired during a run for more extensive characterization of complex protein mixtures.

Table1 Base peak intensity and signal-to-noise ratio(S/N)of proteins in the standard protein mixture from CZE-MS(40-ng proteins)and RPLC-MS(400-ng proteins)
Cyto.c and BSA are not listed in the table because they were not separated well in CZE,and because cyto.c had a very broad peak in RPLC runs.TheS/Nwas estimated via dividing protein intensity by the standard deviation of the background before the protein peaks in the extracted ion electropherogram/chromatogram.We used the most abundant charge state of each protein for peak extraction with a 20-ppm mass tolerance.
2.2 RPLC-MS/MS and CZE-MS/MS for top-down proteomics of E.coli cells
We then applied both RPLC-MS/MS and CZE-MS/MS for top-down MS characterization of anE.coliproteome sample.Two kinds of sample loadings,1-μg and 8-μgE.coliproteins,were tested for RPLC-MS/MS.For CZE-MS/MS,1-μgE.coliproteins was injected for analysis.The same MS and MS/MS parameters were used for CZE-MS/MS and RPLC-MS/MS.
A 240-min total gradient was used for RPLC-MS/MS analyses because the RPLC column was long (90 cm),and the active gradient (separation window)was 160 min.The CZE-MS/MS analysis was completed in 120 min with a separation window of nearly 60 min,as shown in Fig.2a.RPLC-MS/MS produced a much wider separation window than CZE-MS/MS (160 min vs.60 min).CZE-MS/MS with 1-μg proteins produced only 27% lower total ion current (TIC)signal than RPLC-MS/MS with 8-μg proteins (4.03×109vs.5.49×109),and it generated over 4-fold higher TIC signal than RPLC-MS/MS with 1-μg proteins (4.03×109vs.9.26×108).Overall,RPLC-MS/MS identified 245 proteins and 1 004 proteoforms using 8-μg proteins and 105 proteins and 277 proteoforms using 1-μg proteins.CZE-MS/MS identified 159 proteins and 513 proteoforms using 1-μg proteins.A 5% proteoform-level FDR was used to filter the proteoform identifications.
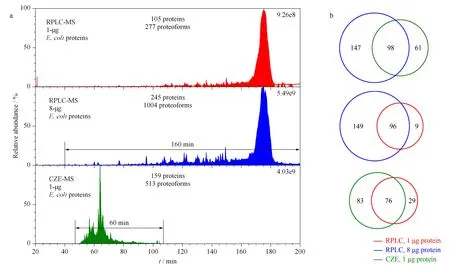
Fig.2 Summary of the E.coli data from RPLC-MS/MS and CZE-MS/MS a.Total ion current (TIC)chromatograms of E.coli proteins from RPLC-MS/MS with 1-μg and 8-μg proteins injected,and the TIC electropherogram of E.coli proteins from CZE-MS/MS with 1-μg proteins injected.b.Protein-level overlaps of RPLC-MS/MS (8-μg proteins),CZE-MS/MS (1-μg proteins),and RPLC-MS/MS (1-μg proteins).
With the same loading amount of 1-μgE.coliproteins,CZE-MS/MS was able to identify 51% more proteins and 85% more proteoforms.However,RPLC-MS/MS with 8-μgE.coliproteins identified 54% more proteins and 96% more proteoforms compared to CZE-MS/MS with 1-μg proteins.This data highlights the advantages and limitations of CZE compared to RPLC.The better sensitivity of CZE results in more protein and proteoform identifications than RPLC-MS/MS with the 1-μg protein material,but the increased loading capacity of RPLC (8-μg proteins)and wider separation window allowed for more protein and proteoform identifications.In order to improve the CZE-MS/MS for more proteoform identifications,we recently developed a CZE-MS/MS system using a much longer LPA-coated capillary compared to this work (1.5 m vs.1 m)[38].The novel CZE-MS/MS system produced a 180-min separation window,leading to the identifications of 800 proteoforms and 260 proteins from anE.colisample [38].
We then evaluated the protein-level overlaps between RPLC-MS/MS and CZE-MS/MS,as shown in Fig.2b.RPLC-MS/MS (8-μg proteins)and CZE-MS/MS (1-μg proteins)identified 306 unique proteins in total with 98 shared between the two methods for a total overlap of 32%.96 of the 105 proteins identified in the 1-μg RPLC-MS/MS run were also found in the 8-μg RPLC-MS/MS run;76 of the 105 proteins identified in the 1-μg RPLC-MS/MS run were identified by CZE-MS/MS.

Fig.4 Sequences and fragmentation patterns of(a)thioredoxin1 with a disulfide bond,(b)Chaperone protein DnaK with an S-glutathionylation,and(c)Chaperone protein DnaK with an S-cysteinylationThe cysteine residues marked with circles have the modifications.
We further compared the RPLC-MS/MS (8-μg proteins)and CZE-MS/MS (1-μg proteins)data regarding theMrof identified proteoforms.The RPLC-MS/MS and CZE-MS/MS runs identified proteoforms withMrin a range of 1-22 kDa and 3-23 kDa,respectively.TheMrdistributions of proteoforms from these two methods have no drastic differences,as shown in Fig.3.We need to note that because the proteoforms identified in this work are relatively small (less than 23 kDa),there may be drastic differences between RPLC-MS/MS and CZE-MS/MS for identification of large proteoforms (larger than 30 kDa),which will be tested in our future work.

Fig.3 Mr distributions of identified proteoforms fromthe E.coli sample using RPLC-MS/MS(8-μg proteins)and CZE-MS/MS(1-μg proteins)
We identified various modifications on the protein sequences,including N-terminal methionine removal,signal peptide cleavage,truncations,N-terminal acetylation,succinylation,disulfide bond,and S-thiolations.For example,we detected a -2-Da mass shift on one proteoform of thioredoxin 1,and the mass shift was localized in a small region of the proteoform sequence (WCGPCK),as shown in Fig.4a,suggesting a disulfide bond between the two cysteine residues.The data agree well with the information in the UniProt database.As another example,we detected two kinds of S-thiolations for Chaperone protein DnaK and they are S-glutathionylation (+305-Da mass shift)and S-cysteinylation (+119-Da mass shift),as shown in Figs.4b and 4c.For the Chaperone protein DnaK proteoforms in Fig.4,the N-terminal methionine was removed,and the C-terminal was truncated.Examples of the identified proteoforms in Fig.4 were from the CZE-MS/MS study.
3 Conclusions
This work represents the first comparison of the state-of-the-art nanoRPLC-MS/MS and CZE-MS/MS for top-down proteomic analysis of a complex proteome.Overall CZE-MS/MS has drastically better sensitivity than nanoRPLC-MS/MS for characterization of a simple intact protein mixture and anE.coliproteome.CZE-MS/MS can be very useful for top-down MS characterization of mass-limited proteome samples.
RPLC has large sample loading capacity and wide separation windows for fraction collection and CZE-MS/MS can characterize proteoforms with high sensitivity.More importantly,RPLC and CZE are orthogonal for separation of proteoforms.Combining RPLC prefractionation to CZE-MS/MS should be an ideal platform for large-scale top-down proteomics.
Acknowledgments
We thank Prof.Heedeok Hong’s group at Michigan State University (Department of Chemistry)for kindly providing theE.colicells for this project.
