甘蓝型油菜开花相关基因的鉴定及进化与表达分析
2019-07-23王艳花曹艳茹李加纳
王艳花 谢 玲 杨 博 曹艳茹 李加纳,*
甘蓝型油菜开花相关基因的鉴定及进化与表达分析
王艳花1,2谢 玲1,2杨 博1,2曹艳茹1,2李加纳1,2,*
1西南大学农学与生物科技学院 / 油菜工程研究中心, 重庆 400715;2西南大学现代农业科学研究院, 重庆 400715
开花是开花植物繁衍后代的前提, 控制开花时间对农作物获得高产稳产具有重要影响。然而, 关于甘蓝型油菜基因组水平上开花相关基因的信息报道较少。本研究对甘蓝型油菜开花相关的基因进行了鉴定、特征分析、进化与表达模式分析。结果表明, 基因组水平上共鉴定到甘蓝型油菜1173个开花相关基因, 这些基因分为9类, 且不均匀地分布在染色体上; 与白菜(AA, 2=20)和甘蓝(CC, 2=18)相比, 甘蓝型油菜(AACC, 2=38)经过自然杂交和染色体加倍后, 使开花相关基因的数目显著扩增; 选择压力分析表明, 糖代谢信号途径的基因比自发途径的开花相关基因受到的选择压力更大; 仍有一些开花相关基因在拟南芥和油菜基因组内非常保守。甘蓝型油菜基因数目的扩增和功能分化导致其开花调控机制更加复杂, 本研究为油菜开花途径提供更多的信息, 也为阐明拟南芥和油菜开花基因的进化关系指明了方向。
甘蓝型油菜; 开花基因; 进化; 调控途径; 表达分析
开花是开花植物成功繁衍的前提。对于农作物而言, 开花时间适当是作物获得高产和优质的重要条件, 而且开花时间受环境影响比较大, 因此, 开花时间是一项重要的农艺指标, 对作物有性繁殖和高产具有重要影响。目前, 大多数影响作物开花时间的遗传组成和环境因子的信息都是从拟南芥研究中获得的[1]。开花时间受外界和自身信号调控[2], 受环境影响的光周期途径和环境温度途径, 受内部信号调控的自主开花途径、赤霉素途径和衰老途径的显著影响[1]。在拟南芥中有超过300个开花相关基因被鉴定(Flowering Interactive Database)[3]。这些基因参与自发途径、光周期、赤霉素、春化和内在因素(包括衰老和糖信号)途径五大开花通路[4]。尽管不同基因参与不同的通路, 但不同通路均受到()、()、()、()等几个关键基因的协同调控[5]。然而, 仅有很少的开花基因在重要作物中得到鉴定。基因组序列的释放和新的计算方法的开发为在基因组水平上鉴定开花相关基因提供了极大便利, 比如在萝卜和小麦中分别鉴定到254个和900个开花相关基因[6]。
油菜是全世界广泛种植的油料作物之一。开花时间在不同油菜品种之间具有非常广泛的变异。前人利用QTL手段研究油菜开花时间, 但只有少数的基因如等得到鉴定或克隆[7-8]。与QTL定位、转录组测序和实验验证方法相比, 比较基因组学成为非常方便的手段来筛选开花候选基因, 特别在非模式作物研究中[6,9], 已经为重要性状进一步的功能验证提供非常重要的信息。目前, 在油菜基因组水平上还没有展开开花相关基因的鉴定工作。本研究在基因组水平上鉴定了油菜开花相关基因, 并揭示了这些基因的进化和表达模式, 为油菜开花研究提供更多的候选基因, 为进一步揭示油菜开花机理奠定基础。
1 材料与方法
1.1 甘蓝型油菜、白菜和甘蓝开花相关基因的筛选
从网站TAIR10 (http://www.arabidopsis.org/)下载拟南芥开花基因的蛋白序列, 从网站BRAD (http:// brassicadb.org/brad/)下载甘蓝、白菜和甘蓝型油菜的基因注释信息。利用拟南芥开花基因的蛋白序列在BRAD网站上进行Blastp分析, 筛选甘蓝型油菜、白菜和甘蓝开花相关基因, 筛选标准为E值 <1e–20、相似度>60%、覆盖率>75%以及匹配长度>70氨基酸残基。
1.2 甘蓝型油菜开花相关基因的染色体分布
从甘蓝型油菜基因组网站(http://www.genoscope. cns.fr/brassicanapus/)下载, 并利用软件Mapchart (https://www.wur.nl/en/show/Mapchart.htm)展示甘蓝型油菜开花相关基因的物理位置信息。
1.3 甘蓝型油菜开花相关基因的选择压力分析
为分析开花相关基因的选择压力, 利用软件PAML计算甘蓝型油菜与拟南芥开花相关基因对的非同义替换率(Ka)与同义替换率(Ks)[10]。当Ka/Ks比值大于1, 等于1或小于1时, 分别表示基因受到正向、中性和纯化选择[11]。
1.4 甘蓝型油菜开花相关基因的表达分析
利用本课题组前期完成的转录组数据(NCBI注册号为PRJNA358784)分析甘蓝型油菜开花相关基因的组织表达情况。利用软件Cufflinks[12]和TopHat2[13]计算基因的表达量FPKM (fragments per kilobase of exon per million mapped reads)。利用软件MeV 4.9.0 (http://en.bio-soft.net/chip/MeV.html)展示表达热图。
2 结果与分析
2.1 甘蓝型油菜开花相关基因的鉴定与染色体分布
利用拟南芥306个开花基因[3],本研究筛选到1173个甘蓝型油菜开花相关基因(附表1),在甘蓝型油菜基因组内可以找到超过89.5%的拟南芥开花基因同源基因(附表2)。大部分的油菜开花相关基因属于光周期、生物钟和光信号途径(图1)。
根据基因的位置信息,1045个开花相关基因被定位在19条染色体上, 剩余128个基因被定位在“unanchored scaffolds”上。这些基因在油菜基因组内分布是非常不均匀的, 在C01上有101个基因,A09上75个,A03上74个,在A08上最少,只有33个基因(图2)。

图1 甘蓝型油菜开花相关基因鉴定与分类
Ag: 衰老途径; At: 环境温度; Au: 自发途径; Cp: 光周期途径; Fd: 花器官发育及顶端分生组织响应途径; Ft: 开花时间调控者; Ho: 激素途径; Su: 糖信号; Ve: 春化。
Ag: aging pathway; At: ambient temperature; Au: autonomous pathway; Cp: clock and photoperiod, pathway; Fd: flower development and apical meristem response pathway; Ft: flowering time integrator; Ho: hormones pathway; Su: sugar signal; Ve: vernalization.
2.2 拟南芥、白菜、甘蓝和甘蓝型油菜开花相关基因的比较分析
在白菜和甘蓝基因组内, 共鉴定出549个和561个开花相关基因(图3和附表3)。根据拟南芥开花基因的分类信息[3], 甘蓝型油菜、白菜和甘蓝的开花同源基因可参与9大类, 即衰老途径、温度、自发途径、光周期、花器官发育、开花调控因子、激素、糖及春化途径。而且, 有些基因可能会参与多个途径, 如和同时参与花器官发育以及开花调控因子。大部分基因属于光周期和自发途径, 温度途径基因在4个物种中均是最少的。
2.3 甘蓝型油菜开花相关基因的关键通路和基因家族分析
2.3.1 光周期途径 光周期是显著影响植物开花时间的外界环境因子[14]。在该途径中, 利用103个拟南芥开花基因, 共鉴定出401个甘蓝型油菜开花相关同源基因(图1和附表1)。作为核心调控因子, CONSTANS(CO)整合外界环境因素如日照长度与内在的生物钟过程来显著调节开花时间[15]。在甘蓝型油菜中共筛选出4个基因, 同时筛选出参与生物钟过程的关键基因如、和[1,16-18]。ZTL、FKF1和LKP2均属于F-box蛋白[19], 但是在甘蓝型油菜基因组内没有筛选到和的同源基因, 而却有6个同源基因。在白菜和甘蓝基因组内同样没有和基因, 表明和的基因丢失以及的三倍化事件同样发生在共同祖先白菜和甘蓝基因组内[20]。在甘蓝型油菜中同样筛选出光信号途径的关键因子如和[1]。
2.3.2 春化和自发途径 许多植物都需要春化过程来诱导开花。大量研究证明()基因和FLOWERING LOCUS C (FLC)基因在春化过程中起至关重要的作用[4]。作为开花抑制因子, FLC被FRI正向调控[21]。在甘蓝型油菜基因组内有9个同源基因。同时, 在甘蓝和白菜基因组内分别检测到2个和1个同源基因, 没有筛选到基因。而且, 很多春化响应基因如、和也在甘蓝、白菜和甘蓝型油菜基因组内被检测到。与春化途径相似, 自发途径基因如()、()、、和通过抑制的表达来间接诱导开花[22-23]。在甘蓝型油菜基因组内, 有461个基因属于自发途径。

(图2)

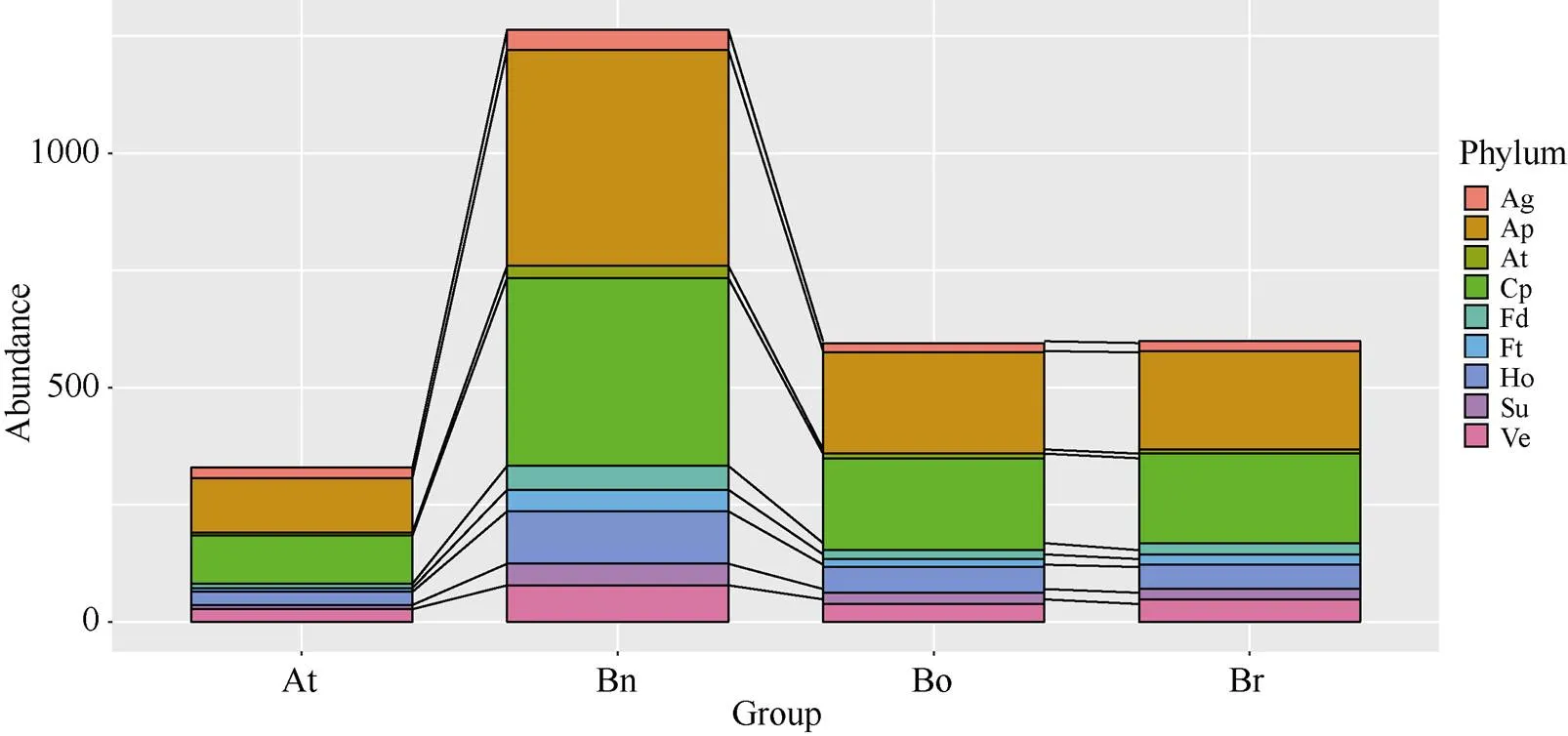
图3 不同开花途径基因在拟南芥、白菜、甘蓝和甘蓝型油菜基因组内的数目
缩写同图1。Abbreviation are the same as those given in Fig. 1.
2.3.3 顶端分生组织响应途径 开花时间受到复杂的遗传因子的调控, 这类调控因子被称之为“调控者”基因, 可以整合内外环境信号和不同途径的信号[24-25]。在本研究中, 91个开花相关基因被鉴定为“调控者”基因, 比如等。在甘蓝型油菜基因组内仅有1个同源基因。
2.3.4 赤霉素信号与代谢途径 很多证据表明, 在短日照情况下, 赤霉素是开花的必要条件, 但在长日照条件下则不需要。截至目前, 在拟南芥中有28个基因参与赤霉素途径, 这些基因主要分为赤霉素合成途径基因如和[26]和信号传导因子基因如和[27]。在甘蓝型油菜基因组内, 除之外, 所有赤霉素相关基因都有同源基因。GAI属于DELLA蛋白家族, 但另外一个DELLA蛋白家族成员在甘蓝型油菜基因组内却有7个同源基因, 该基因抑制植物从苗期向花期的转变[28]。
2.3.5 温度途径 温度可以显著影响开花时间[29]。适当高温可以促进开花, 在拟南芥中已经有响应温度的关键基因被鉴定, 如、和[1]。在甘蓝型油菜中, 筛选到6个同源基因, 该基因在高温环境中通过结合的启动子来诱导开花[30]。作为MADS-box蛋白, SVP通过响应外界温度来调节开花时间, 该蛋白通过结合在和的启动子区域来抑制开花[31], 而EFM通过正调节SVP来参与调控开花时间[32]。在甘蓝型油菜中,和各有4个同源基因。
2.3.6 糖信号途径 除了赤霉素, 糖也可以参与调节开花时间[1]。特别指出的是, 蔗糖和海藻糖显著影响开花时间[33-34]。在甘蓝型油菜中, 46个影响开花的糖代谢相关的同源基因被鉴定。作为正向调控开花因子, TREHALOSE-6-PHOSPHATE SYNTHASE 1 (TPS1)参与海藻糖合成, 不管长日照还是短日照环境, 下调该基因都将导致开花延后[35]。编码锌指类转录因子, 通过调控糖的运输和代谢来促进开花[36]。
2.3.7 衰老途径 在甘蓝型油菜基因组内共鉴定出43个影响开花的衰老途径同源基因。该途径的关键基因包括(和)、和[1]。很多证据表明miRNAs参与开花调控。其中和分别编码miR156和miR172。随着植物年龄增长, miR156表达量下调, 但是miR172表达量升高。miR156和miR172分别通过调控靶基因和基因来调控开花时间。作为开花抑制因子, 不管长日照还是短日照, 过表达SMZ都可以延迟开花[37-38]。miR156的靶基因通过诱导miR172来间接促进开花[39]。另外, HXK1参与果糖和葡萄糖的磷酸化, 其通过抑制miR156间接促进开花, 突变体在长日照环境中推迟开花时间[40]。
2.4 开花相关基因的选择压力分析
同源基因对的Ka/Ks比值被用来当作选择压力的指标。对于开花相关基因在拟南芥和油菜之间的垂直同源基因对的选择压力, 计算表明, 各开花相关基因通路的Ka/Ks比值范围为0.18~0.46 (图4和附表4), 表明开花相关基因受到纯化选择压力。
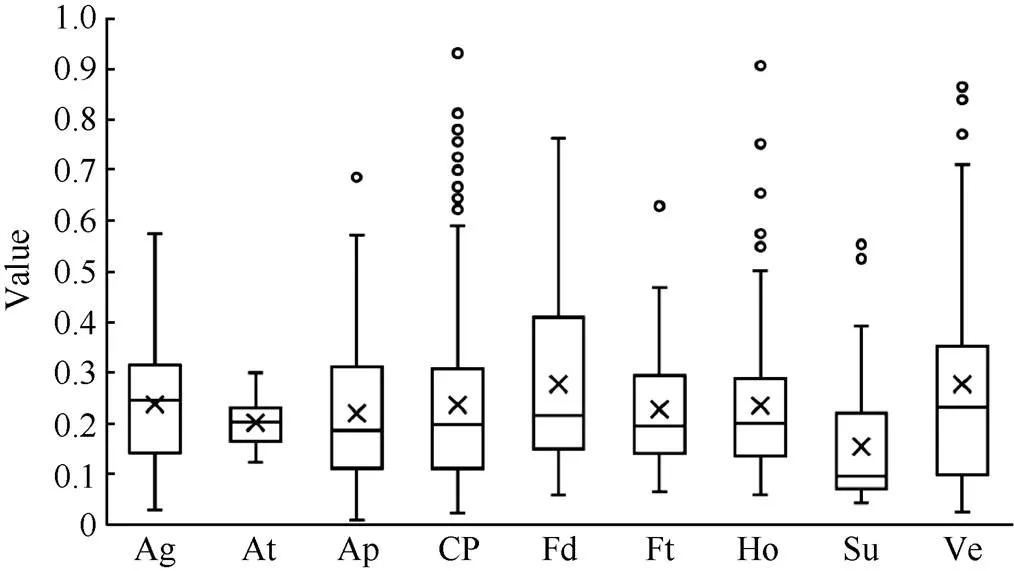
图4 甘蓝型油菜不同开花途径基因的选择压力分析
缩写同图1。Abbreviations are the same as those given in Fig. 1.
2.5 甘蓝型油菜开花相关基因的表达分析
本研究利用课题组前期完成的以“中双11”为材料获得的各时期各组织表达的转录组数据(NCBI注册号为PRJNA358784)分析甘蓝型油菜开花基因的表达特征。由图5可知, 非功能化、新功能化或次功能化现象在重复基因对内出现, 尽管多数基因对具有相似的表达模式, 但有些具有新的功能。
3 讨论
白菜、甘蓝和甘蓝型油菜基因组数据的释放为研究重要农艺性状的基因及功能提供了极大的便利[41-43]。虽然很多基因在拟南芥中都已经研究的很透彻, 但其同源基因在以上3种近源物种中的功能尚不明确。为补充这些信息, 本研究利用拟南芥开花相关基因, 通过同源比对的方法筛选到1173个甘蓝型油菜、561个甘蓝和549个白菜开花相关同源基因。考虑到白菜、甘蓝和甘蓝型油菜的基因组大小, 甘蓝型油菜基因组内的开花相关基因的数目比其在白菜和甘蓝基因组内数量的总和还要多[41,43]。甘蓝型油菜基因组内的开花相关基因数目是其在拟南芥基因组内的3.8倍, 有些同源基因数目超过6个, 有些则低于3个, 表明同源基因内发生了丢失、重组、功能保留和分化。然而, 这并不影响甘蓝型油菜主要的开花途径, 因为油菜同样可以整合环境内外的信号来调控开花时间。
在甘蓝型油菜中筛选到一些光周期途径的关键调控因子基因, 如、和。且在甘蓝型油菜基因组内筛选到2个同源基因, 这2个成员在各组织器官中的表达量均较高。在甘蓝型油菜中有7个同源基因, 且该基因在拟南芥中已经得到验证[44], 其中4个成员表达量较高, 而其余3个成员几乎不表达, 可能发生非功能化。在甘蓝型油菜中筛选得到的6个同源基因在所有检测的组织中表达量均较高, 可能诱导的表达[45]。生物钟途径的关键调控因子的2个同源基因也同样具有较高的表达量[17]。在甘蓝型油菜中有4个同源基因, 其中2个表达量较高, 而其余2个几乎不表达。GI和CO的存在表明在甘蓝型油菜中GI/CO调控开花时间的方式是非常保守的。相比其他途径基因, 光周期途径的基因的Ka/Ks比值的变异范围更广。
作为开花关键调控因子, 在甘蓝型油菜基因组内分别检测到9个、4个、6个、3个、4个和5个。在自发和春化途径中, FLC是关键的调控因子, 其通过与FRI互作来抑制开花过程[46]。尽管在甘蓝型油菜基因组内有9个同源基因, 但其表达量都很低, 可能跟取材时间有关。在甘蓝型油菜中鉴定得到自发途径和长日照条件下起重要作用的4个同源基因[47], 但只有1个成员表达量较高。整合光周期、春化和自发途径的关键调控因子[48]在甘蓝型油菜中有6个同源基因, 其中4个表达量较高。编码一个促进早开花的转录因子, 该转录因子与APETALA1 (AP1)协同作用[49], 在甘蓝型油菜中有4个成员, 其中2个在主序顶端中高表达, 5个同源基因, 均在花蕾、花萼、萼片、花瓣和主序顶端中高表达。总之, 几个开花关键调控因子可以通过整合不同途径信号来调控开花时间, 表明开花调控过程在拟南芥和油菜中是非常保守的。

图5 甘蓝型油菜开花相关基因的组织表达分析
缩写同图1。Abbreviations are the same as those given in Fig. 1.
4 结论
在甘蓝型油菜中鉴定出1173个开花相关基因, 并分为9大类。开花调控机制在拟南芥和油菜之间是非常保守的, 保留了主要的调控通路和调节因子。光周期途径比其他途径的选择压力变异更大。这为甘蓝型油菜开花调控机制提供更多的信息, 为育种家培育新品种提供基础。
附表 请见网络版: 1) 本刊网站http://zwxb.chinacrops.org/; 2) 中国知网http://www.cnki.net/; 3) 万方数据http://c.wanfangdata.com.cn/Periodical-zuowxb. aspx。
[1] Srikanth A, Schmid M. Regulation of flowering time: all roads lead to Rome., 2011, 68: 2013–2037.
[2] Koornneef M, Alonso B C, Peeters A J M, Soppe W. Genetic control of flowering time in Arabidopsis., 1998, 49: 345–370.
[3] Bouche F, Lobet G, Tocquin P, Perilleux C. FLOR-ID: an interactive database of flowering-time gene networks in., 2016, 44: 1167–1171.
[4] Blumel M, Dally N, Jung C. Flowering time regulation in crops-what did we learn from Arabidopsis?., 2015, 32: 121–129.
[5] Fornara F, de Montaigu A, Coupland G. SnapShot: control of flowering in Arabidopsis., 2010, 141: 550–550.
[6] Peng F Y, Hu Z, Yang R C. Genome-wide comparative analysis of flowering-related genes in Arabidopsis, wheat, and barley., 2015, 15: 1–17.
[7] Raman H, Raman R, Coombes N, Song J, Prangnell R, Bandaranayake C, Tahira R, Sundaramoorthi V, Killian A, Meng J, Dennis E S, Balasubramanian S. Genome-wide association analyses reveal complex genetic architecture underlying natural variation for flowering time in canola., 2016, 39: 1228–1239.
[8] Xu L, Hu K, Zhang Z, Guan C, Chen S, Hua W, Li J, Wen J, Yi B, Shen J, Ma C, Tu J, Fu T. Genome-wide association study reveals the genetic architecture of flowering time in rapeseed (L.)., 2016, 23: 43–52.
[9] Wang J, Qiu Y, Cheng F, Chen X, Zhang X, Wang H, Song J, Duan M, Yang H, Li X. Genome-wide identification, characterization, and evolutionary analysis of flowering genes in radish (L.)., 2017, 18: 981, doi: 10.1186/s12864-017-4377-z.
[10] Yang Z. PAML 4: phylogenetic analysis by maximum likelihood., 2007, 24: 1586–1591.
[11] Liang Y, Wan N, Cheng Z, Mo Y, Liu B, Liu H, Raboanatahiry N, Yin Y, Li M. Whole-genome identification and expression pattern of the vicinal oxygen chelate family in rapeseed (L.)., 2017, 8: 745, doi: 10.3389/fpls.2017.00745.
[12] Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley D R, Pimentel H, Salzberg S L, Rinn J L, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks., 2012, 7: 562–578.
[13] Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg S L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions., 2013, 14: R36.
[14] Song Y H, Ito S, Imaizumi T. Flowering time regulation: photoperiod- and temperature-sensing in leaves., 2013, 18: 575–583.
[15] Wenkel S, Turck F, Singer K, Gissot L, Le Gourrierec J, Samach A, Coupland G. CONSTANS and the CCAAT box binding complex share a functionally important domain and interact to regulate flowering of Arabidopsis., 2006, 18: 2971–2984.
[16] Dong M A, Farre E M, Thomashow M F. Circadian clock-associated 1 and late elongated hypocotyl regulate expression of the C-repeat binding factor (CBF) pathway in Arabidopsis., 2011, 108: 7241–7246.
[17] Sawa M, Kay S A. GIGANTEA directly activates flowering locus T in., 2011, 108: 11698–11703.
[18] Nagel D H, Doherty C J, Pruneda P J L, Schmitz R J, Ecker J R, Kay S A. Genome-wide identification of CCA1 targets uncovers an expanded clock network in Arabidopsis., 2015, 112: 4802–4810.
[19] Baudry A, Ito S, Song Y H, Strait A A, Kiba T, Lu S, Henriques R, Pruneda P J L, Chua N H, Tobin E M, Kay S A, Imaizumi T. F-box proteins FKF1 and LKP2 act in concert with ZEITLUPE to control Arabidopsis clock progression., 2010, 22: 606–622.
[20] Lou P, Wu J, Cheng F, Cressman L G, Wang X, McClung C R. Preferential retention of circadian clock genes during diploidization following whole genome triplication in., 2012, 24: 2415–2426.
[21] Johanson U, West J, Lister C, Michaels S, Amasino R, Dean C. Molecular analysis of FRIGIDA, a major determinant of natural variation in Arabidopsis flowering time., 2000, 290: 344–347.
[22] Noh B, Lee S H, Kim H J, Yi G, Shin E A, Lee M, Jung K J, Doyle M R, Amasino R M, Noh Y S. Divergent roles of a pair of homologous jumonji/zinc-finger-class transcription factor proteins in the regulation of Arabidopsis flowering time., 2004, 16: 2601–2613.
[23] Simpson G G. The autonomous pathway: epigenetic and post- transcriptional gene regulation in the control of Arabidopsis flowering time., 2004, 7: 570–574.
[24] Parcy F. Flowering: a time for integration., 2005, 49: 585–593.
[25] Kim S, Soltis P S, Wall K, Soltis D E. Phylogeny and domain evolution in the APETALA2-like gene family., 2006, 23:107–120.
[26] Mitchum M G, Yamaguchi S, Hanada A, Kuwahara A, Yoshioka Y, Kato T, Tabata S, Kamiya Y, Sun T P. Distinct and overlapping roles of two gibberellin 3-oxidases in Arabidopsis development., 2006, 45: 804–818.
[27] Ariizumi T, Murase K, Sun T P, Steber C M. Proteolysis- independent downregulation of DELLA repression in Arabidopsis by the gibberellin receptor GIBBERELLIN INSENSITIVE DWARF1., 2008, 20: 2447–2459.
[28] Achard P, Vriezen W H, Van D S D, Harberd N P. Ethylene regulates arabidopsis development via the modulation of DELLA protein growth repressor function., 2003, 15: 2816–2825.
[29] Samach A, Wigge P A. Ambient temperature perception in plants., 2005, 8: 483–486.
[30] Thines B C, Youn Y, Duarte M I, Harmon F G. The time of day effects of warm temperature on flowering time involve PIF4 and PIF5., 2014, 65: 1141–1151.
[31] Lee J H, Yoo S J, Park S H, Hwang I, Lee J S, Ahn J H. Role of SVP in the control of flowering time by ambient temperature in Arabidopsis., 2007, 21: 397–402.
[32] Yan Y, Shen L, Chen Y, Bao S, Thong Z, Yu H. A MYB-domain protein EFM mediates flowering responses to environmental cues in Arabidopsis., 2014, 30: 437–448.
[33] Paul M J, Primavesi L F, Jhurreea D, Zhang Y. Trehalose metabolism and signaling., 2008, 59: 417–441.
[34] Dalchau N, Baek S J, Briggs H M, Robertson F C, Dodd A N, Gardner M J, Stancombe M A, Haydon M J, Stan G B, Goncalves J M, Webb A A. The circadian oscillator genemediates a long-term response of thecircadian clock to sucrose., 2011, 108: 5104–5109.
[35] Wahl V, Ponnu J, Schlereth A, Arrivault S, Langenecker T, Franke A, Feil R, Lunn J E, Stitt M, Schmid M. Regulation of flowering by trehalose-6-phosphate signaling in., 2013, 339: 704–707.
[36] Seo P J, Ryu J, Kang S K, Park C M. Modulation of sugar metabolism by an INDETERMINATE DOMAIN transcription factor contributes to photoperiodic flowering in Arabidopsis., 2011, 65: 418–429.
[37] Jung J H, Seo Y H, Seo P J, Reyes J L, Yun J, Chua N H, Park C M. The GIGANTEA-regulated microRNA172 mediates photoperiodic flowering independent of CONSTANS in Arabidopsis., 2007, 19: 2736–2748.
[38] Mathieu J, Yant L J, Murdter F, Kuttner F, Schmid M. Repression of flowering by the miR172 target SMZ., 2009, 7: e1000148.
[39] Wang J W, Czech B, Weigel D. miR156-regulated SPL transcription factors define an endogenous flowering pathway in, 2009, 138: 738–749.
[40] Matsoukas I G, Massiah A J, Thomas B. Starch metabolism and antiflorigenic signals modulate the juvenile-to-adult phase transition in Arabidopsis., 2013, 36: 1802–1811.
[41] Wang X, Wang H, Wang J, Sun R, Wu J, Liu S, Bai Y, Mun J H, Bancroft I, Cheng F, Huang S, Li X, Hua W, Wang J, Wang X, Freeling M, Pires J C, Paterson A H, Chalhoub B, Wang B, Hayward A, Sharpe A G, Park B S, Weisshaar B, Liu B, Li B, Liu B, Tong C, Song C, Duran C, Peng C, Geng C, Koh C, Lin C, Edwards D, Mu D, Shen D, Soumpourou E, Li F, Fraser F, Conant G, Lassalle G, King G J, Bonnema G, Tang H, Wang H, Belcram H, Zhou H, Hirakawa H, Abe H, Guo H, Wang H, Jin H, Parkin I A, Batley J, Kim J S, Just J, Li J, Xu J, Deng J, Kim J A, Li J, Yu J, Meng J, Wang J, Min J, Poulain J, Wang J, Hatakeyama K, Wu K, Wang L, Fang L, Trick M, Links M G, Zhao M, Jin M, Ramchiary N, Drou N, Berkman P J, Cai Q, Huang Q, Li R, Tabata S, Cheng S, Zhang S, Zhang S, Huang S, Sato S, Sun S, Kwon S J, Choi S R, Lee T H, Fan W, Zhao X, Tan X, Xu X, Wang Y, Qiu Y, Yin Y, Li Y, Du Y, Liao Y, Lim Y, Narusaka Y, Wang Y, Wang Z, Li Z, Wang Z, Xiong Z, Zhang Z,Genome Sequencing Project C. The genome of the mesopolyploid crop species., 2011, 43: 1035–1039.
[42] Chalhoub B, Denoeud F, Liu S, Parkin I A, Tang H, Wang X, Chiquet J, Belcram H, Tong C, Samans B, Correa M, Da S C, Just J, Falentin C, Koh C S, Le C I, Bernard M, Bento P, Noel B, Labadie K, Alberti A, Charles M, Arnaud D, Guo H, Daviaud C, Alamery S, Jabbari K, Zhao M, Edger P P, Chelaifa H, Tack D, Lassalle G, Mestiri I, Schnel N, Le P M C, Fan G, Renault V, Bayer P E, Golicz A A, Manoli S, Lee T H, Thi V H, Chalabi S, Hu Q, Fan C, Tollenaere R, Lu Y, Battail C, Shen J, Sidebottom C H, Wang X, Canaguier A, Chauveau A, Berard A, Deniot G, Guan M, Liu Z, Sun F, Lim Y P, Lyons E, Town C D, Bancroft I, Wang X, Meng J, Ma J, Pires J C, King G J, Brunel D, Delourme R, Renard M, Aury J M, Adams K L, Batley J, Snowdon R J, Tost J, Edwards D, Zhou Y, Hua W, Sharpe A G, Paterson A H, Guan C, Wincker P. Early allopolyploid evolution in the post-Neolithicoilseed genome., 2014, 345: 950–953.
[43] Liu S, Liu Y, Yang X, Tong C, Edwards D, Parkin I A, Zhao M, Ma J, Yu J, Huang S, Wang X, Wang J, Lu K, Fang Z, Bancroft I, Yang T J, Hu Q, Wang X, Yue Z, Li H, Yang L, Wu J, Zhou Q, Wang W, King G J, Pires J C, Lu C, Wu Z, Sampath P, Wang Z, Guo H, Pan S, Yang L, Min J, Zhang D, Jin D, Li W, Belcram H, Tu J, Guan M, Qi C, Du D, Li J, Jiang L, Batley J, Sharpe A G, Park B S, Ruperao P, Cheng F, Waminal N E, Huang Y, Dong C, Wang L, Li J, Hu Z, Zhuang M, Huang Y, Huang J, Shi J, Mei D, Liu J, Lee T H, Wang J, Jin H, Li Z, Li X, Zhang J, Xiao L, Zhou Y, Liu Z, Liu X, Qin R, Tang X, Liu W, Wang Y, Zhang Y, Lee J, Kim H H, Denoeud F, Xu X, Liang X, Hua W, Wang X, Wang J, Chalhoub B, Paterson A H. Thegenome reveals the asymmetrical evolution of polyploid genomes., 2014, 5: 3930, doi: 10.1038/ncomms4930.
[44] Fujiwara S, Oda A, Yoshida R, Niinuma K, Miyata K, Tomozoe Y, Tajima T, Nakagawa M, Hayashi K, Coupland G, Mizoguchi T. Circadian clock proteins LHY and CCA1 regulate SVP protein accumulation to control flowering in Arabidopsis., 2008, 20: 2960–2971.
[45] Ding Z, Millar A J, Davis A M, Davis S J. TIME FOR COFFEE encodes a nuclear regulator in thecircadian clock., 2007, 19: 1522–1536.
[46] Tao Z, Shen L, Liu C, Liu L, Yan Y, Yu H. Genome-wide identification of SOC1 and SVP targets during the floral transition in Arabidopsis., 2012, 70: 549–561.
[47] Wigge P A, Kim M C, Jaeger K E, Busch W, Schmid M, Lohmann J U, Weigel D. Integration of spatial and temporal information during floral induction in Arabidopsis., 2005, 309: 1056–1059.
[48] Lee H, Suh S S, Park E, Cho E, Ahn J H, Kim S G, Lee J S, Kwon Y M, Lee I. The AGAMOUS-LIKE 20 MADS domain protein integrates floral inductive pathways in Arabidopsis., 2000, 14: 2366–2376.
[49] Liu C, Xi W, Shen L, Tan C, Yu H. Regulation of floral patterning by flowering time genes., 2009, 16: 711–722.
Flowering genes in oilseed rape: identification, characterization, evolutionary and expression analysis
WANG Yan-Hua1,2, XIE Ling1,2, YANG Bo1,2, CAO Yan-Ru1,2, and LI Jia-Na1,2,*
1College of Agronomy and Biotechnology, Southwest University/ Chongqing Engineering Research Center for Rapeseed, Chongqing 400715, China;2Academy of Agricultural Sciences, Southwest University, Chongqing 400715, China
Flowering is a prerequisite for successful sexual reproduction. Controlling of flowering time is important for crop production in different geographical regions. However, few information regarding flowering genes or their evolution at genome-wide level inhas been reported. In this study, identification, characterization, evolutionary and expression analysis of flowering genes in oilseed rape were performed. In total, 1173 flowering-related genes classified into nine types and distributed unevenly on the chromosomes were identified at the genome level of. Compared with(AA, 2= 20) and(CC, 2= 18),(AACC, 2= 38) showed significantly enlarge number of flowering-related genes due to natural hybridization and chromosome doubling. Selective pressure analysis showed that the autonomous pathway genes had less selectionpressure than the genes involved in sugar metabolic pathway, suggesting that some key flowering-related genes are relatively conserved betweenand. The present study provides more information on theflowering pathways and sheds light on the evolutionary relationship of flowering-related genes betweenand.
; flowering genes; evolution; regulatory pathway;expression analysis
2018-11-16;
2019-04-15;
2019-05-08.
10.3724/SP.J.1006.2019.84159
李加纳, E-mail: ljn1950@swu.edu.cn, Tel: 023-68250642
E-mail: hawer313@163.com
本研究由重庆市民生工程主题专项项目(cstc2016shms-ztzx80020)和高等学校学科创新引智基地项目(“111”项目)(B12006)资助。
This study was supported by the Special Project of Chongqing People’s Livelihood Project (cstc2016shms-ztzx80020) and the Project of Intellectual Base for Discipline Innovation in Colleges and Universities (“111” Project) (B12006).
URL:http://kns.cnki.net/kcms/detail/11.1809.S.20190505.1436.008.html
