Protective potential of glucagon like peptide 2 (GLP-2) against the neurodegeneration
2019-07-18AntonellaAmato,FlaviaMulè
Neurodegeneration consists in loss of neuron specific types, pattern and distribution, leading to progressive dysfunctions of the central nervous system. Neurodegenerative diseases include diverse pathological conditions, among which Alzheimer's and Parkinson's diseases are the most prevalent ones. Alzheimer's disease is known as a growing dementia, characterized by progressive language, memory, and cognitive loss,while Parkinson's disease is primarily characterized as a motor disorder.Senile plaques, caused by amyloid β peptide, hyperphosphorylated taubased neurofibrillary tangles and synapse loss, are the principal pathological hallmarks of Alzheimer's disease. Amyloid β oligomer formation is associated with development of reactive oxygen and nitrogen species,inflammation, calcium-dependent excitotoxicity, impairment of cellular respiration, and alteration of synaptic functions related with learning and memory. Parkinson's disease is produced by dopaminergic neuron deterioration in the extrapyramidal tract of the midbrain. Accumulation of α-synuclein proteins (Lewy bodies) in the central, autonomic,and peripheral nervous system is the hallmark of the Parkinson's disease. The Levy bodies break the neuronal membrane leading to neuronal death through oxidative stress, excitotoxicity, energy failure and neuroinflammation.
Factors associated with the development of neurodegenerative diseases:The development of neurodegenerative diseases may result from a number of factors including aging, genetics and environmental factors. In fact, at the cellular level, aging is associated with accumulating oxidative stress, declining mitochondrial function, impaired DNA repair and decreased tissue regeneration. As mitochondrial efficiency declines with age, the mitochondrial dysfunction may contribute to the formation of misfolded protein aggregates and subsequently may lead to Alzheimer's and Parkinson's diseases. In addition, neuronal death, due to activated microglia and astrocytes producing reactive oxygen species and pro-inflammatory cytokines, further will promote the activation of glial cells, developing a neurotoxic vicious cycle of inflammation that perpetuates tissue injury. However, the etiology of the neurodegenerative diseases is multifactorial. External factors, including lifestyle and diet, are linked with the risk of neurodegeneration. Metabolic diseases, such as obesity and type 2 diabetes have been correlated with an increased probability of developing cognitive decline. Indeed,insulin has a significant role in modulation of synaptic plasticity and learning memory, then it plays a role in maintaining cognitive functions. Alterations in the concentration of insulin and in the insulin receptor expression have been reported in Alzheimer's disease brains,suggesting that irregularities in the insulin-signaling pathway may contribute to impairment of memory function. Indeed, several evidences suggest that a high fat diet (HFD) can lead to obesity, insulin resistance and dementia. In HFD mice, the obesity is associated with peripheral and central insulin resistance, cerebral inflammation, increase in oxidative stress and expression of amyloid β precursor protein, amyloid β,beta-secretase 1 and glycogen synthase kinase 3β (Nuzzo et al., 2015),enzymes involved in amyloid β precursor protein processing and formation of amyloid plaques and neurofibrillary tangles, respectively.Therefore, HFD mice are used as a model to study the effects of potential beneficial factors for neurodegeneration, because they show in the cortex increased markers of Alzheimer's disease, formation of plaques as well as impaired spatial learning (Valladolid-Acebes et al., 2011;Nuzzo et al., 2015; Wu et al., 2018). Vascular pathology also contributes to neurodegenerative disorders and vascular risk factors aggravate cognitive impairment in different chronic neurodegenerative disease. Loss of brain-blood barrier integrity results in increased vascular permeability and it is associated with reduced cerebral blood flow and impaired haemodynamic responses. Breakdown of the brain-blood barrier enables toxic blood-derived molecules, cells and microbial agents to enter the brain, triggering inflammatory and immune responses, involved in the neurodegeneration. Continuously reduced blood supply in brains contributes to a decrease in brain metabolism and causes a series of endogenous neuronal changes. Chronic cerebral hypoperfusion (CCH)has been shown to potentiate amyloid pathology and to induce microglia activation, and blood-brain barrier disruption. In CCH, autophagy signaling is activated, provoking neuroinflammation and oxidative stress, amyloid β protein accumulation and tau hyperphosphorylation,with cognition impairment (Back et al., 2017).
Glucagon like peptide 2 (GLP-2) and neurodegeneration:Recently significant decrease in hippocampal levels of GLP-2 receptor (GLP-2R) has been reported after CCH in rats, suggesting that this may be related to cognitive impairment induced by blood low perfusion (Xie et al., 2018). Moreover, upregulation of GLP-2R expression can rescue the cognitive impairment induced by CCH (Xie et al., 2018). This finding together with other recent evidence lead to consider GLP-2 as a protective and improving memory factor. Indeed, GLP-2 had no effect on memory function in normal mice, but the peptide protects and improves memory function in mice injected with intracerebroventricular lipopolysaccharides, which induces neuroinflammation and pro-inflammatory cytokine expression increase (Iwai et al., 2015). Moreover,injections of GLP-2 improve the learning and memory ability in mice with vascular dementia (Chi et al., 2017). GLP-2 is a 33-amino-acid peptide, which is synthesized not only in L-cells of the small intestine and colon, but also in a discrete population of neurons in the brainstem and hypothalamus. The GLP-2R, a G protein-coupled receptor, which is widely expressed in the gut, has been localized also within the central nervous system including the hypothalamus, hippocampus and cortex.Although different biological actions have been described in the gut,the role of GLP-2 in the central nervous system is less known, because few researches investigated the role of GLP-2 in the nervous system.The first studies in vitro suggested that the GLP-2R activation protects neurons from excitotoxic damage, because it is able to reduce the glutamate-induced cell death in cultured hippocampal cells (Lovshin et al.,2004) and it acts as a trophic factor for astrocytes, because it enhances the proliferation of these cells (Velázquez et al., 2009). In vivo studies suggested that the central administration of GLP-2 to rodents decreased gut motility and blood pressure, suppressed food intake, suggesting that the peptide may act as a satiation signal to the hypothalamus, and produced antidepressant-like effects. Moreover, intracerebroventricular infusion of GLP-2 increased glucose tolerance and insulin sensitivity by activating GLP-2R and intracellular phosphatidylinositol 3-kinase-dependent signaling pathways in proopiomelanocortin expressing neurons (Shi et al., 2013) (Figure 1). Indeed, GLP-2 may act as a protective factor against the deregulation of the glucose metabolism that occurs in obese conditions, at least in animal model (Baldassano et al., 2015),condition which could lead to neurodegeneration (Nuzzo et al., 2015).Mice lacking GLP-2R selectively in hypothalamic arcuate nucleus proopiomelanocortin neurons display impaired postprandial glucose tolerance and hepatic insulin resistance suggesting a GLP-2 physiological role in glycaemic control. The GLP-2 beneficial action on central insulin sensitivity could be beneficial for neurodegeneration because when impairment of insulin signaling occurs, protein kinase B (Akt) is down-phosphorylated and Foxo3a translocates from the cytoplasm to the nucleus where it activates its pro-apototic target genes. A variety of other biological activities, however, could be interpreted as suggesting a neuroprotective role in the brain (Figure 1). In cultured hippocampal neurons GLP-2R activation acutely and dose-dependently stimulates de novo synthesis of protein by the phosphatidylinositol 3-kinase-dependent Akt-mammalian target of rapamycin (mTOR) signaling pathway(Shi et al., 2012). mTOR is a key downstream effector of phosphatidylinositol 3-kinase-dependent Akt, involved in the regulation of cell growth, proliferation, and survival, protein synthesis, and angiogenesis.Therefore, GLP-2-induced stimulation of protein synthesis might be physiologically relevant to providing secondary mediators (neuropeptides or growth factors) and to maintaining neuronal survival. Xie et al.(2018) analyzed not only how GLP2-R is involved in the development of learning and memory impairment caused by CCH, but also the underlying mechanisms. The results from their experiments suggest that GLP-2R over-expression can improve the impairment of long-term potentiation and long-term depression, and consequently the cognitive damage induced by CCH. GLP-2R downregulation may be involved in CCH-induced cognitive impairment by inhibiting synaptic plasticity via decreased AKT-mTOR signaling. This phosphatidylinositol 3-kinase-AKT-mTOR-p70S6K pathway is involved in synapse-related protein synthesis, synaptic plasticity, long-term potentiation, long-term depression, neurogenesis, and memory formation in hippocampus.Indeed, GLP-2R increases the neurogenesis in the dentate gyrus, neuronal activity, and density of dendritic spines and mushroom spines in hippocampal neurons (Figure 1). Further recent direct evidences for a protective and beneficial role in the brain have been reported in the animal model (Figure 1) (Nuzzo et al., 2019). GLP-2 is able to exert anti-inflammatory and anti-oxidative activities in the central nervous system. Brains of HFD mice, treated for four weeks with the stable analogue of the GLP-2, [Gly2]-GLP-2 (teduglutide), show reductions in several inflammation and stress markers (nuclear factor-kappa B,interleukin-8, tumor necrosis factor-α, interleukin-1β and interleukin-6, phosphorylated extracellular regulated protein kinases, heat shock protein 60, inducible nitric oxide synthase) as well as the reactive oxygen species genesis in comparison with the brain of HFD-untreated mice. Moreover, [Gly2]-GLP-2 inhibits the increased expression of GFAP, considered as index of gliosis and glia activation, observed in superficial and deep areas of the cortex of HFD mice. Therefore, the peptide could minimize the deleterious inflammatory effects induced by HFD. In addition, [Gly2]-GLP-2 reduces the presence of neurons with fragmented DNA in brain cortex sections. In fact, the increase in the cell number with fragmented DNA, index of an impairment of cell survival, which is present in the cortical superficial and deep areas of the HFD untreated-animals, has not been observed in the [Gly2]-GLP-2-treated HFD mice, suggesting a preventive role of the peptide in inducing apoptosis (Nuzzo et al., 2019) (Figure 1). [Gly2]-GLP-2 treatment significantly reduces the HFD-induced increase in the expression of amyloid β precursor protein without modifying the expression levels of beta-secretase 1 and PSN1, enzymes involved in amyloid β precursor protein processing, suggesting that these enzymes are not involved in amyloid β precursor protein processing yet, likely due to the age of mice used for the experiments. Therefore, whether endogenous or exogenous central nervous system GLP-2R signaling modulates actions specifically involved in Alzheimer's disease development requires additional investigation.
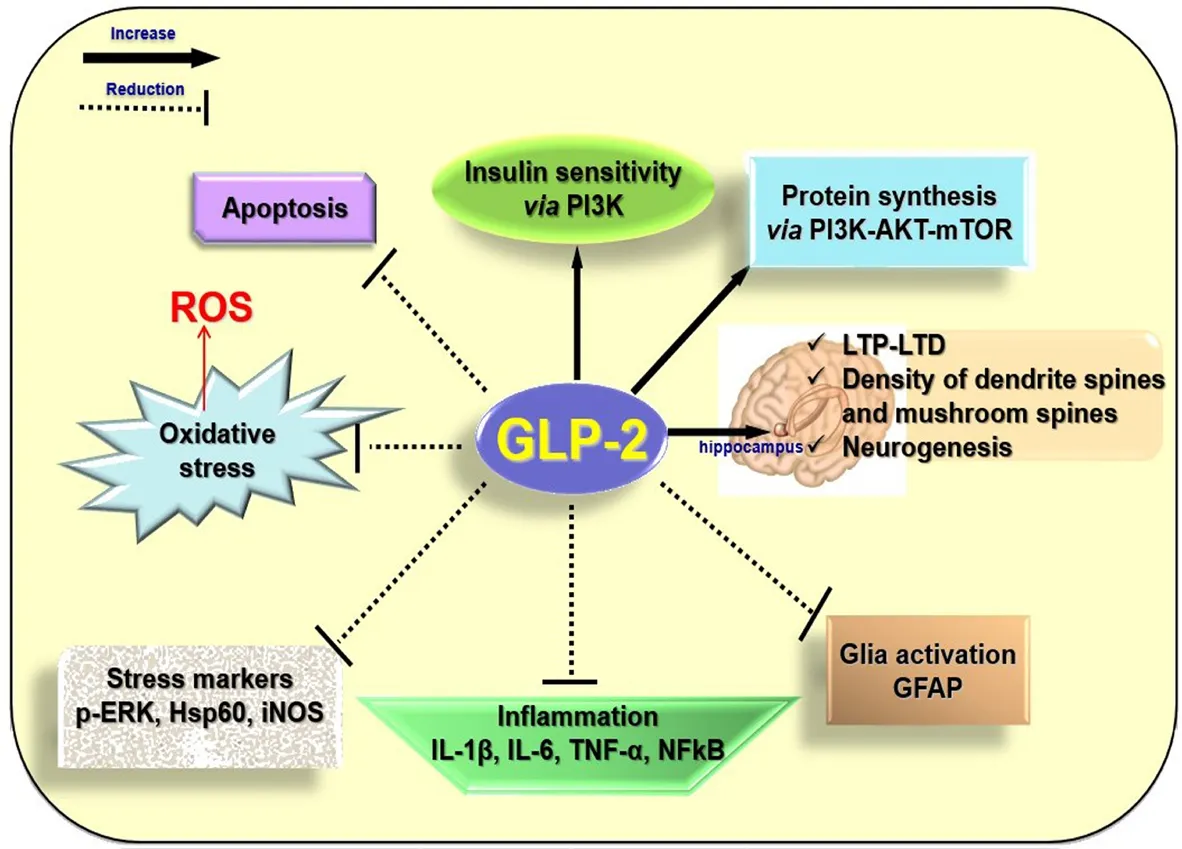
Figure 1 Putative actions responsible for the beneficial effects of glucagon like peptide 2 (GLP-2) in the neurodegeneration.Exogenous GLP-2 is able to reduce the expression of markers of inflammation and stress, the glia activation and the apoptosis in the brains of high fat diet mice. Intracerebroventricular infusion of GLP-2 increases glucose tolerance and insulin sensitivity by activating GLP-2 receptor (GLP-2R) and intracellular phosphatidylinositol 3-kinase (PI3K)-dependent signaling pathways in proopiomelanocortin expressing neurons. In cultured hippocampal neurons, GLP-2R activation acutely and dose-dependently stimulates de novo synthesis of protein by the phosphatidylinositol 3-kinase-dependent protein kinase B (Akt)-mammalian target of rapamycin (mTOR) signaling pathway. GLP-2R over-expression improves the impairment of long-term potentiation (LTP) and long-term depression (LTD) and it increases the neurogenesis in the dentate gyrus, neuronal activity, and density of dendritic spines and mushroom spines in hippocampal neurons. p-ERK:Phosphorylated extracellular regulated protein kinases; Hsp60:heat shock protein 60; iNOS: inducible nitric oxide synthase; IL:interleukin; TNF-α: tumor necrosis factor-α; NFkB: nuclear factor-kappa B; GFAP: glial fibrillary acidic protein; ROS: reactive oxygen species.
Conclusion and perspectives:Although recent evidence underlines the importance of GLP-2 and GLP-2R in neuroprotection in animal model,we are still far from having an in depth understanding of the complex neurobiology of GLP-2. At this time, much remains to be done to characterize fully the functional roles and signaling events of GLP-2 in the brain. In particular, additional studies are required to validate and define the peptide involvement under neurodegenerative conditions.These studies may shed light on new approaches to promote neuronal regeneration and repair following injury or disease.
Antonella Amato, Flavia Mulè*Dipartimento di Scienze e Tecnologie Biologiche Chimiche e Farmaceutiche (STEBICEF) Università di Palermo, Palermo, Italy
*Correspondence to: Flavia Mulè, PhD, flavia.mule@unipa.it.
orcid: 0000-0003-0676-0142 (Flavia Mulè)
Received: February 5, 2019
Accepted: April 25, 2019
doi: 10.4103/1673-5374.259612
Copyright license agreement: The Copyright License Agreement has been signed by both authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:Sandra I. Mota, Center for Neurosciences and Cell Biology, Portugal.
Additional file:Open peer review report 1.
杂志排行

中国神经再生研究(英文版)的其它文章
- Etomidate affects the anti-oxidant pathway to protect retinal ganglion cells after optic nerve transection
- Pain inhibition through transplantation of fetal neuronal progenitors into the injured spinal cord in rats
- MicroRNA expression in the hippocampal CA1 region under deep hypothermic circulatory arrest
- Atsttrin reduces lipopolysaccharide-induced neuroinflammation by inhibiting the nuclear factor kappa B signaling pathway
- Association between PPARG genetic polymorphisms and ischemic stroke risk in a northern Chinese Han population: a case-control study
- Silencing Huwe1 reduces apoptosis of cortical neurons exposed to oxygen-glucose deprivation and reperfusion