Identification of microRNAs and messenger RNAs involved in human umbilical cord mesenchymal stem cell treatment of ischemic cerebral infarction using integrated bioinformatics analysis
2019-07-17YinMengQuXinSunXiuLiYanHangJinZhenNiGuoYiYang
Yin-Meng Qu, Xin Sun, Xiu-Li Yan, Hang Jin, Zhen-Ni Guo , Yi Yang,
1 Stroke Center, Neuroscience Center, Department of Neurology, the First Hospital of Jilin University, Changchun, Jilin Province, China
2 Clinical Trial and Research Center for Stroke, Department of Neurology, the First Hospital of Jilin University, Changchun, Jilin Province, China
Abstract In recent years, a large number of differentially expressed genes have been identified in human umbilical cord mesenchymal stem cell(hUMSC) transplants for the treatment of ischemic cerebral infarction. These genes are involved in various biochemical processes, but the role of microRNAs (miRNAs) in this process is still unclear. From the Gene Expression Omnibus (GEO) database, we downloaded two microarray datasets for GSE78731 (messenger RNA (mRNA) profile) and GSE97532 (miRNA profile). The differentially expressed genes screened were compared between the hUMSC group and the middle cerebral artery occlusion group. Gene ontology enrichment and pathway enrichment analyses were subsequently conducted using the online Database for Annotation, Visualization, and Integrated Discovery. Identified genes were applied to perform weighted gene co-suppression analyses, to establish a weighted co-expression network model. Furthermore, the protein-protein interaction network for differentially expressed genes from turquoise modules was built using Cytoscape (version 3.40) and the most highly correlated subnetwork was extracted from the protein-protein interaction network using the MCODE plugin. The predicted target genes for differentially expressed miRNAs were also identified using the online database starBase v3.0. A total of 3698 differentially expressed genes were identified. Gene ontology analysis demonstrated that differentially expressed genes that are related to hUMSC treatment of ischemic cerebral infarction are involved in endocytosis and inflammatory responses. We identified 12 differentially expressed miRNAs in middle cerebral artery occlusion rats after hUMSC treatment, and these differentially expressed miRNAs were mainly involved in signaling in inflammatory pathways, such as in the regulation of neutrophil migration. In conclusion, we have identified a number of differentially expressed genes and differentially expressed mRNAs, miRNA-mRNAs, and signaling pathways involved in the hUMSC treatment of ischemic cerebral infarction. Bioinformatics and interaction analyses can provide novel clues for further research into hUMSC treatment of ischemic cerebral infarction.
Key Words: nerve regeneration; ischemic cerebral infarction; human umbilical cord mesenchymal stem cell treatment; bioinformatics analysis;differentially expressed genes; differentially expressed mRNAs; inflammatory response; stem cell therapy; weighted gene co-suppression analysis;WGCNA; protein-protein interaction network; PPI; hUMSC; neural regeneration
Graphical Abstract
Introduction
In recent years, intravenous thrombolysis and vascular interventions have made great progress for the clinical therapy of ischemic stroke, but stroke remains one of the leading causes of death worldwide (Lin et al., 2011). Existing therapies mainly focus on improving collateral circulation and increasing the expression of neurological factors, but currently there are no therapies that can fundamentally repair damaged nerve tissue (Zhang et al., 2016). Numerous experimental studies have confirmed that stem cell transplantation for ischemic stroke can achieve cell replacement and functional reconstruction, and can also promote nerve repair,with promising prospects for clinical application (Adams et al., 2007; Lee et al., 2010; Bernstock et al., 2017). Stem cells,including mesenchymal stem cells, endothelial progenitor cells, and neural stem cells, are currently used to treat ischemic stroke. Mesenchymal stem cells are the most commonly used stem cells; these are mainly derived from bone marrow,umbilical cord, and adipose tissue (Chen et al., 2016). Human umbilical cord mesenchymal stem cells (hUMSCs) are advantageous because they have convenient materials, come from a wide variety of sources, have no ethical controversy,and cause weak immune rejection. They are therefore the ideal stem cell type for the treatment of ischemic stroke (Lu et al., 2006; Duya et al., 2013). However, the mechanisms of hUMSC treatment for ischemic stroke, and the best route for hUMSC transplantation, are not still clear.
MicroRNAs (miRNAs) are broadly distributed, highly conserved, non-coding endogenous small RNA molecules that are approximately 18 to 25 nucleotides in length. Based on complementation with the sequence of the target messenger RNA (mRNA), miRNAs can degrade mRNAs and inhibit protein translation, thereby affecting gene expression (Liu et al., 2010). MiRNAs and their whip mRNAs form complex regulatory networks that participate in a series of crucial life processes, such as cell growth, differentiation, proliferation,migration, death, stress response, and cellular inflammation and immunity. They are also related to the occurrence and development of major diseases, such as vascular diseases,blood system diseases, nervous system diseases, and cancers(Rink and Khanna, 2011; Selvamani et al., 2012). In recent years, a variety of miRNAs have been identified to be associated with ischemic stroke. For example, miR-497 exerts anti-apoptotic effects via the inhibition of Bcl-2 and Bcl-w gene expression during the pathogenesis of ischemic stroke(Sun et al., 2013). Animal experiments have revealed that miRNAs in ischemic brain tissue can be significantly up- or down-regulated, suggesting that miRNA may be involved in the regulation of injury and mechanisms of protection after cerebral ischemia (Zhai et al., 2012). The roles of miRNAs in cerebral ischemia may include involvement in post-ischemic cell apoptosis and differentiation, vascular inflammatory responses, ischemia and hypoxia, and ischemia and reperfusion, thereby affecting the development and prognosis of ischemic injury (Duan et al., 2014).
Bioinformatics analyses of gene profile microarrays have been widely used to identify important molecular mechanisms and biomarkers in various diseases. In recent years,many studies have been conducted investigating differentially expressed genes (DEGs) in the hUMSC treatment of ischemic cerebral infarction, and it has been reported that a large number of genes take part in molecular functions and biological processes and pathways (Guo et al., 2017; Zhai et al., 2017). However, uncertainty remains concerning the molecular pathways between DEGs and miRNAs in the mechanism of hUMSC treatment of ischemic cerebral infarction.This study aimed to use bioinformatics to identify the interactions between DEGs and differentially expressed miRNAs(DEMs), together with their potential mechanisms, in the hUMSC treatment of ischemic cerebral infarction.
Materials and Methods
Microarray data
Two microarray datasets, GSE78731 (mRNA profile) and GSE97532 (miRNA profile), of middle cerebral artery occlusion (MCAO) rats were obtained from the Gene Expression Omnibus (GEO) database (https://www. ncbi.nlm.nih.gov/geo/). GSE78731 contained 16 Sprague-Dawley (SD) rat samples based on the Agilent-028279 SurePrint G3 Rat GE 8x60K Microarray (Probe Name version, https://www.selectscience.net/products/sureprint-g3-rat-ge-8-x-60k-microarray/?prodid=113098) platform, in which RNA was isolated from the ipsilateral hemisphere in rats without MCAO (normal group, n = 5), as well as in rats treated with an intravenous injection of 1 × 106hUMSC (hUMSC group, n = 6) and saline (MCAO group, n = 5) at 24 hours post-MCAO. For the GSE97532 dataset, six SD rats were subjected to MCAO(n = 3) or a sham operation (n = 3). The miRNA expression in the blood was compared between the two groups using an Affymetrix Multispecies miRNA-4 Array platform.
Identification of differentially expressed genes
Background correction and quartile data normalization of the downloaded data were performed using a robust multi-array averaging algorithm (Katz et al., 2006). Probes that did not contain corresponding gene symbols were neglected, and the mean value of gene symbols containing multiple probes was calculated. All identified DEGs between these two groups were screened using a Student's t-test and fold change (FC) test using the Limma package in R software (https://www.r-project.org/). Using a threshold of P values < 0.05 and FC absolute values > 2, volcano plots were run using the R software ggplot 2 package to identify DEGs that had statistically significant differences between the two groups. Hierarchical clustering and comprehensive analysis of DEGs were then carried out.
Gene function analysis
We searched the National Center for Biotechnology Information (NCBI) gene database based on the following search terms: “rat” in order to detect any relationship between rat and DEGs. The Database for Annotation, Visualization, and Integrated Discovery (DAVID) (http://david.abcc.ncifcrf.gov/) was used to enrich the target gene ontology (GO) analysis. Genes in the GO term (including cellular components,biological processes, and molecular functions) with P values below 0.05 were considered to be significantly enriched. The Kyoto Encyclopedia of Genes and Genomes (KEGG) is a database resource (http://www.genome.jp/kegg/) that clarifies the effects and advanced functions of biological systems.This resource was used identify significant differences in DEGs or target genes in KEGG pathways. Next, we used Cytoscape (version 3.40, https://cytoscape.org/) to visualize the relationship between DEGs and biological processes.
Weighted gene co-suppression analysis
Samples were clustered to investigate the relationships between clinical features and expression profiles. After pre-processing, the raw data was significantly identified by the genetic module according to the weighted gene co-suppression analysis (WGCNA) algorithm package in R software. The probe set was first filtered based on the variance value of the expression levels in all samples. Probe sets with overlapping gene symbols were removed based on expression variation in WGCNA. In summary, the individual correlation coeffi-cients of the paired selected genes were calculated to obtain a similarity matrix (sij). Next, we converted the matrix into an adjacency matrix (aij) using the power function. Modules of tightly interconnected genes were identified by performing average chain hierarchical clustering. Genes that were not assigned to a particular module were designated in gray.
Analysis and construction of a protein-protein interaction network and subnetwork
An online database protein-protein interaction (PPI) network was built using STRING (https://string-db.org/). First,the PPI network consists of pairs of PPIs, and chooses a larger score. Next, Cytoscape software was used to visualize the interaction relationships between genes and analyze them using their topological properties by computing network parameters (including network degree distribution) using the CentiScaPe application. Finally, a subnetwork was extracted from the PPI network using the MCODE application.
Predicting miRNA-mRNA interactions
The regulatory relationship between DEGs and DEMs was predicted using starBase (http://starbase.sysu.edu.cn/). A visualization of miRNA-DEG networks was performed using Cytoscape.
Results
Identification of differentially expressed mRNAs in human umbilical cord mesenchymal stem cell treatment of ischemic cerebral infarction
First, the data in the GEO database (GSE78731) were clustered and the samples were divided into normal, MCAO,and hUMSC groups, as shown in Figure 1A. However,cluster analysis showed that MCAO1 and MCAO5 were classified into the normal group, so we discarded these two samples. Normal2, Normal3, and hUMSC3-5 were also discarded based on similar principles, leaving a total of three groups consisting of nine samples. A cluster analysis of these nine samples revealed that they could be divided into the aforementioned three groups of samples (Figure 1B). We then identified 2149 DEGs between the MCAO and normal groups (Figure 1C), 289 DEGs between the hUMSC and MCAO groups (Figure 1D), and 3371 DEGs between the hUMSC and normal groups (Figure 1E). All DEGs are shown in the Venn diagram (Figure 1F). We then used these differentially expressed mRNAs to construct a network for hUMSC treatment of ischemic cerebral infarction.
Functional and pathway enrichment analyses of differentially expressed genes in human umbilical cord mesenchymal stem cell treatment of ischemic cerebral infarction
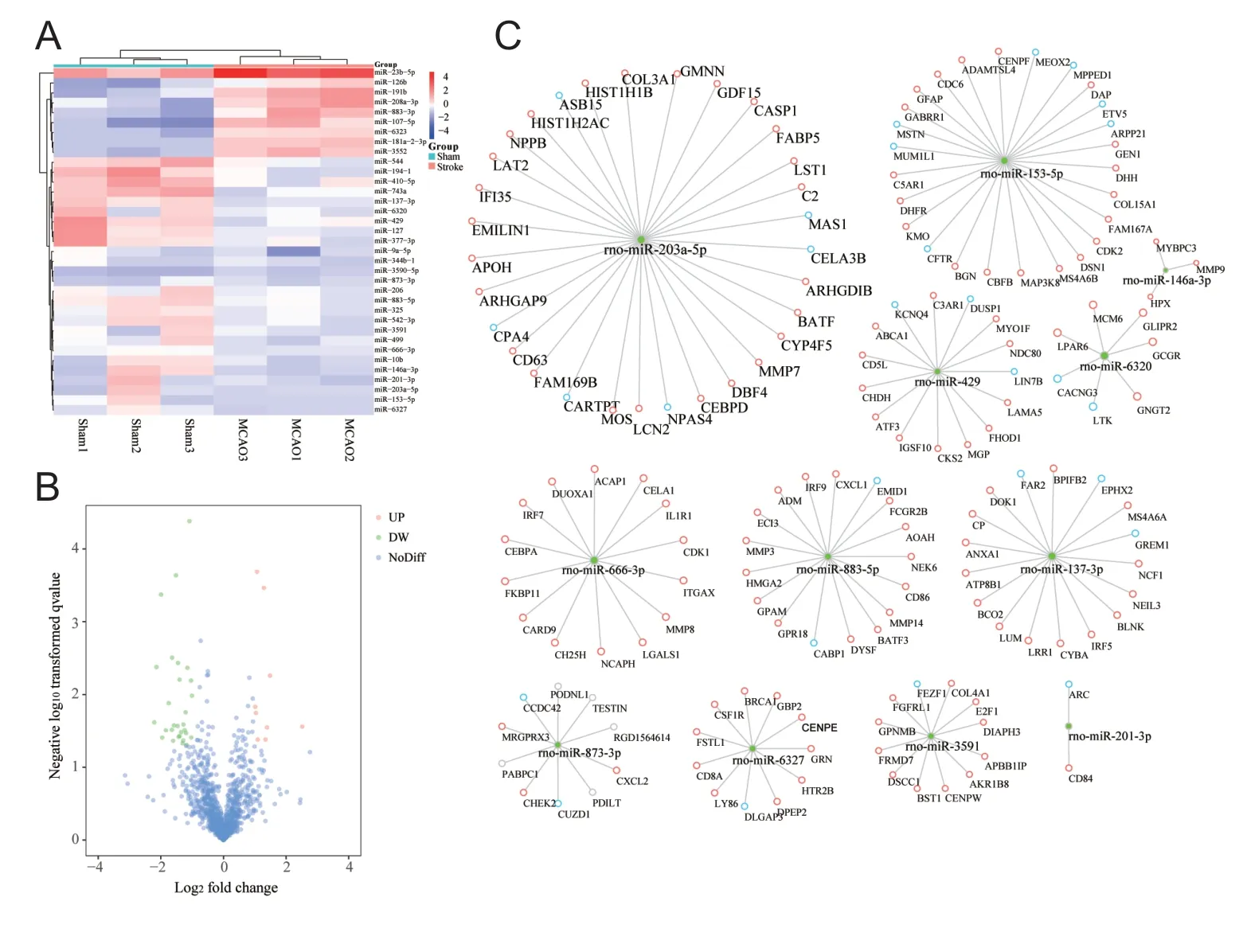
We matched the unique genes among the three groups and screened out 3698 DEGs. A heatmap of DEG expression is shown in Figure 2A. DAVID was used to explore the functions and pathways that DEGs were mainly enriched in. GO enrichment analysis indicated that, in the MCAO group,altered signaling pathways included a significant decrease in the retinoic acid-inducible gene-I-like receptor signaling pathway, the Fc epsilon RI signaling pathway, and inflammatory mediator regulation of the transient receptor potential channels signaling pathway; in terms of biological processes, these genes were mainly involved in the inflammatory response, response to drugs, and response to lipopolysaccharides (Figure 2B & C). These results suggest that inflammatory factor-mediated apoptosis or necrosis may be an important mechanism for MCAO development or hUMSC therapy.
Protein-protein interaction network construction and analysis of modules
To further explore the potential targets and common mechanisms of MCAO progression and hUMSC therapy, we performed a correlation analysis of gene expression and disease characteristics using WGCNA. Cluster and soft threshold analysis (Figure 3A & B) and gene expression modules (Figure 3C) gave characteristic maps of two key factors (stroke and hUMSC). In the four modules, the gene expression of the turquoise-colored module was most correlated with these two factors (r = 0.8 or 0.65), so we selected the genes from this module for subsequent analysis. Figure 4 shows the network created by importing these genes into the STRING database. The 12 highest-scoring nodes were screened as central genes: Rab13, LON687121, Kifc1, Ttk, C1qa, Ube2c,Ccl3, Rps20, RT1-DMb, Gins1, Plk1, and Tax1bp3. The network showed the subnetwork analysis by MCODE, and all the modules were down-regulated.
Pairing of miRNAs with differentially expressed genes
We analyzed the profile obtained under accession number GSM97532 to identify DEMs between the two sets of samples, and screened 33 DEMs. The volcano plot of these DEMs is shown in Figure 5A. Twenty-seven miRNAs were down-regulated in animals with ischemic cerebral infarction (Figure 5B). Comparing the target genes to DEGs from GSE97532, we screened miRNAs with opposite expression trends for further analysis and predicted target genes using starBase v3.0 (Figure 5C). We established 12 miRNA-mRNA regulatory networks involved in the pathogenesis of stroke,from which miRNAs and target genes can be selected for validation. We identified that, in the MCAO model, the most important changes in miRNA expression in blood components relative to the normal group were in miRNA-203a-5p and miRNA-153-5p. This result suggests that these two miRNAs may play an important role in stroke pathogenesis.
The miRNA-mRNA regulatory network in signaling pathways
As shown in Figure 6, corresponding signaling pathways in ischemic cerebral infarction were analyzed using the ClueGo plugin. These pathways were mainly related to the immune system, such as in the regulation of neutrophil migration.Other enriched pathways included the nucleotide-binding oligomerization domain-like receptor signaling pathway,granulocyte chemotaxis, the hydrogen peroxide metabolic process, the integrin-mediated CD63 signaling pathway, and the regulation of pathway-restricted SMAD protein phosphorylation.
Discussion
HUMSCs can promote the recovery of neurological function in an animal model of ischemic brain injury, and hUMSC therapy is expected to become a novel treatment for ischemic brain injury in humans (Zhang et al., 2017). In addition, hUMSCs may play an active role in the treatment of ischemic brain injury through neuronal protection, remodeling of neural circuits, endogenous angiogenesis, regulation of immune inflammatory responses, and regulation of neurometabolites (Sun et al., 2017). However, the exact mechanism by which hUMSCs play this role is not yet clear.
In the present study, Rab13, which had the highest degree in the PPI network, was associated with the endocytosis pathway. Increasing evidence has confirmed that Rab13 is highly expressed in the lungs, kidney, and whole brain and spinal cord, but its subcellular localization in these tissues remains unclear (Novick and Zerial, 1997). Zahraoui et al.(1994) reported that Rab13 is localized to tight junctions in polar epithelial cells. The Rab gene codes for a small group of GTPases that regulate and control membrane communication between cells and organelles, and Rab protein is involved in many key processes of endocytosis and membrane circulation. When establishing and maintaining the polarity of cells such as epithelial cells, a variety of Rab proteins are involved in the regulation of membrane transport, and Rab13 is one of them. However, its relationship with cerebral ischemia has not been reported.
With continuing scientific research, the correlation between miRNA and the occurrence and development of cerebral ischemia and its specific pathogenesis will be gradually clarified; this may provide novel insights for the diagnosis,treatment, and prognosis evaluation of cerebral ischemia(Sorensen et al., 2014). MiRNAs are involved in inflammatory reactions, ion balance, and oxidative stress following cerebral ischemia. Following focal cerebral ischemia and cerebral ischemia-reperfusion, there are wide and continuous changes in miRNA expression in brain tissue (Dharap et al., 2009; Chi et al., 2014). It has been reported that miR-146a may play a neuroprotective role following permanent MCAO in adult Wistar rats by regulating the Toll-like receptor signaling pathway in ischemic cortical epithelial cells(Zhang et al., 2012). In addition, the expression of miR-15a and miR-497 is elevated and the expression of anti-apoptotic factor Bcl-2 is down-regulated in the cerebral cortex of the ischemic hemisphere in adult SD rats at 72 hours after reperfusion. Moreover, silencing of miR-15a and miR-497 by antisense oligonucleotides injected into the lateral ventricle may play a neuroprotective role by up-regulating the expression of Bcl-2 (Yin et al., 2010, 2012; Shi et al., 2013). In our study,12 DEMs were identified as down-regulated in ischemic cerebral infarction, including miR-230-5p, miR-153-5p, miR-666-3p, and miR-883-5p. Previous studies have confirmed that the expression of miR-145, miR-21, miR-24, and others change in ischemic cerebral infarction (Jia et al., 2015; Liu et al., 2016). Therefore, the miRNAs that we screened and their targeted genes may also play critical roles in ischemic cerebral infarction abnormalities.
Although many studies have reported potential biomarkers for stroke, none of them are similar to the miRNAs that were identified in the present study. The reason for this discrepancy may be a result of the variability in these studies. For example, previous reported research used different species:humans and rodents (Chen et al., 2017). Although miRNAs are believed to be stable across different species, this may be an important factor in the reported variation. In addition,the present study has some limitations because the two sets of data are derived from different types of tissues; GSE97532 used blood samples while GSE78731 used brain samples.However, extracellular miRNAs have been observed to have extraordinary stability in various body fluids. By studying the relationship between serum miRNA and disease, it was reported that blood miRNAs may be ideal molecular biomarkers for the diagnosis and treatment of diseases. In addition, tissue cells also actively release miRNA into the blood circulatory system. For example, many tumor-specific miRNAs are detected in the circulatory system at different stages of disease development, and Ng et al. (2009) reported that miRNAs in plasma or serum reflect trends observed in tumor-specific miRNAs. Therefore, we believe that it is not only reasonable to compare mRNA in tissue with miRNA in blood, but also to use these to screen for MCAO biomarkers.
Our study identified DEGs using bioinformatics analyses and identified potential miRNAs to predetermine the regulatory mechanisms of ischemic cerebral infarction. We screened 3698 DEGs and 12 DEMs that may play a crucial role in the development of hUMSC treatment of ischemic cerebral infarction. The miRNA-mRNA regulatory network may provide novel insights for future studies of the mechanisms of ischemic cerebral infarction. However, further experimental studies are required to validate the results from these bioinformatics analysis.
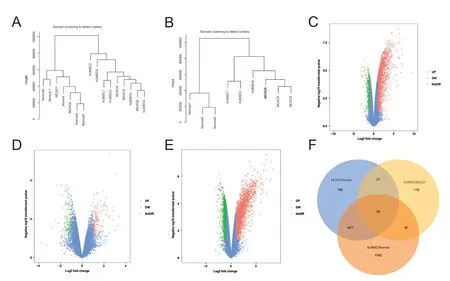
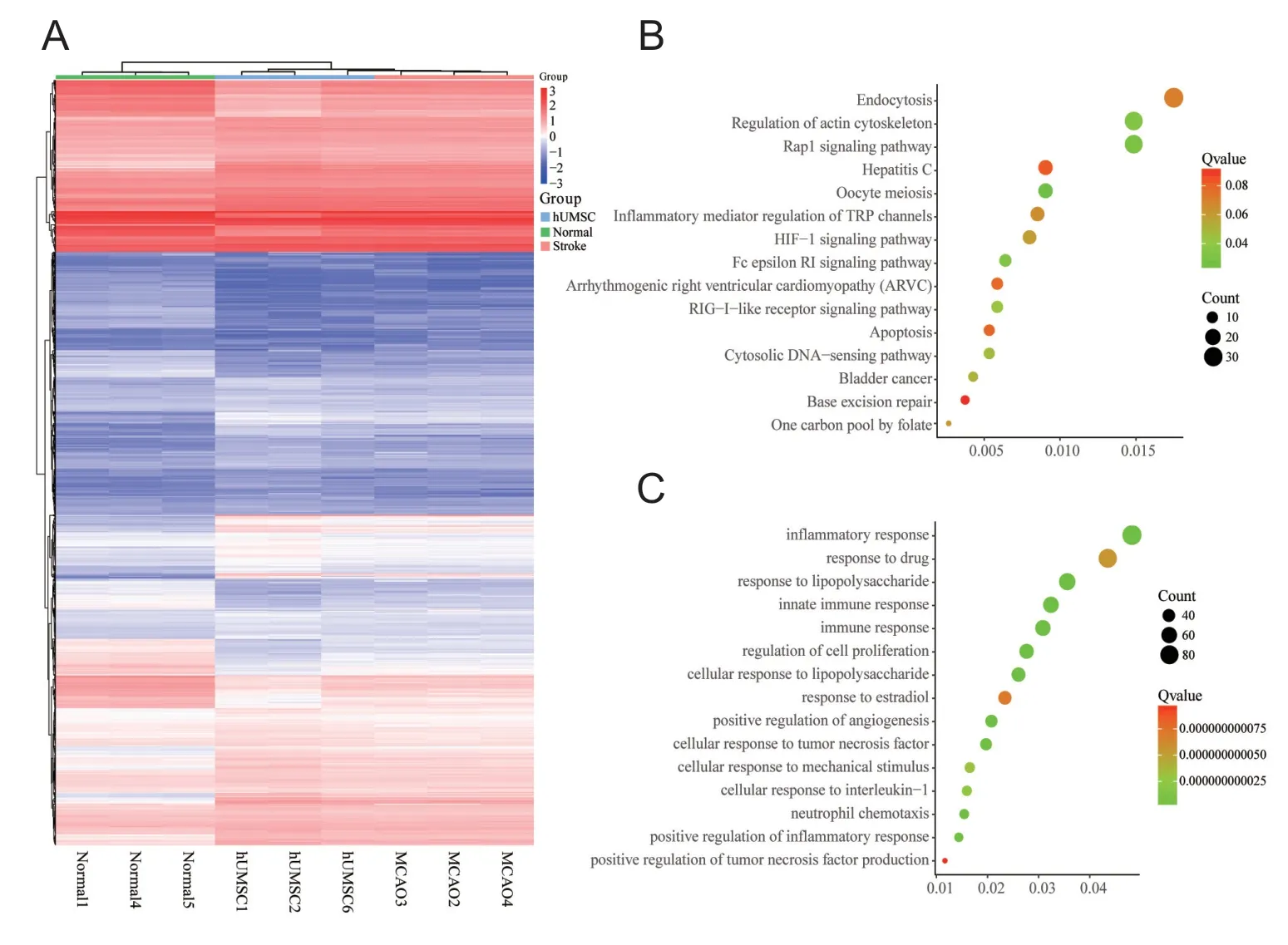
Figure 2 Functional and pathway enrichment analyses in middle cerebral artery occlusion (MCAO)from GSE78731 (mRNA profile).(A) Heat map of the expression of 3698 differentially expressed genes(DEGs); red indicates up-regulated genes and blue indicates down-regulated genes. (B, C) The signal pathway enrichment analysis (B) and biological process (BP) enrichment analysis (C) of mRNA data. The first 15 significantly enriched pathways,biological processes, and their scores(Count and Q value) were shown on the x and y axes, respectively. TRP:Transient receptor potential; HIF-1:hypoxia-inducible factor-1; RIG: retinoic acid-inducible gene; hUMSC:human umbilical cord mesenchymal stem cell.
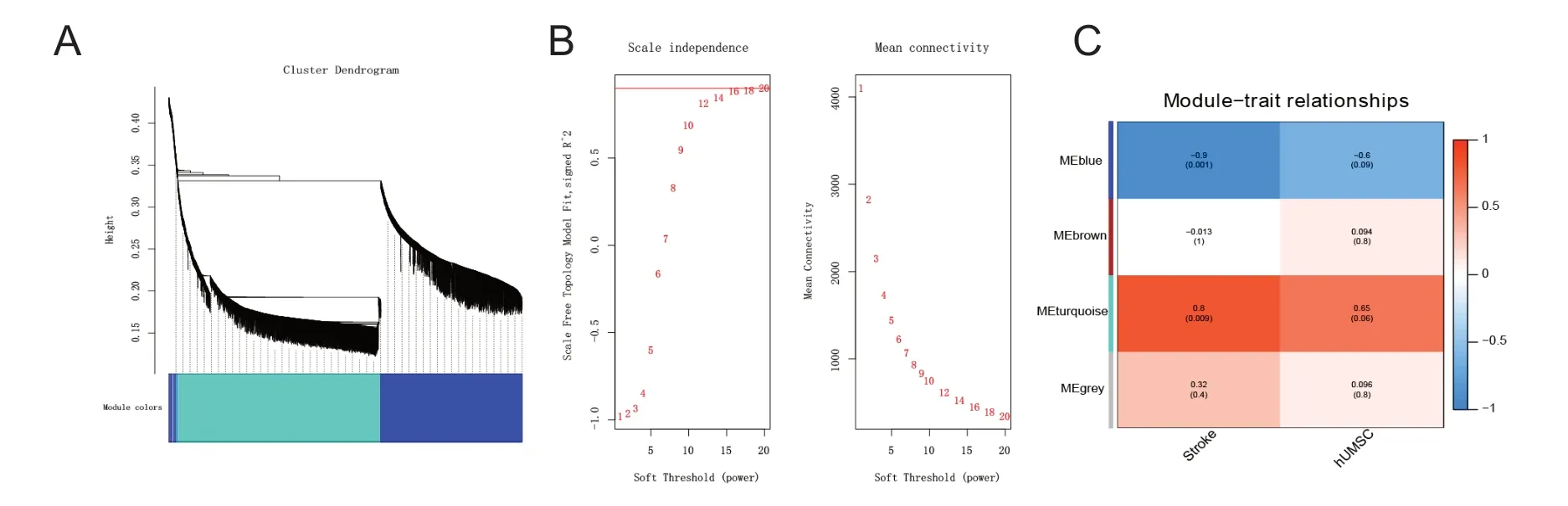
Figure 3 Identification of hub genes from GSE78731 (mRNA profile) by weighted gene co-suppression analysis (WGCNA).(A) Cluster dendrogram based on dynamic tree cut: the clustering maps and modules identified by the weighted gene co-expression network analysis show clustering maps of different colors. (B) Figures for soft threshold. Left: Scale independence; right: mean connectivity. (C) The feature quantity matrix of modules. The number of rows is the number of modules to be filtered, and the number of columns is the number of samples.
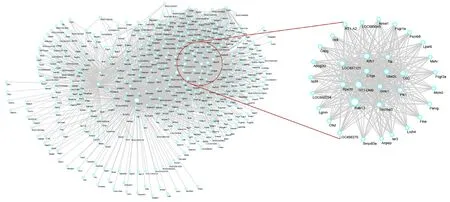
Figure 4 Protein-proteininteraction network construction and analysis in the turquoisemodule.A search tool was used to search the STRING database (https://string-db.org/), and differentially expressed genes (DEGs) from the turquoise module were filtered into a DEG protein-protein interaction network. The areas in the circle are statistically significant modules.The subnetwork consisted of 12 nodes and 23 edges.
Author contributions:Study implementation, data analysis and manuscript writing: YMQ; study concept and design: YY, ZNG; technical and information support: XS, XLY, HJ. All authors approved the final version of the paper.
Conflicts of interest: None declared.
Financial support:This study was supported by the National Key Research & Development Program of China, No. 2016YFC1301600, and Program for Jilin University Science and Technology Innovation Team, No.2017TD-12 (both to YY). The funding bodies played no role in the study design, in the collection, analysis and interpretation of data, in the writing of the paper, and in the decision to submit the paper for publication.
Institutional review board statement: No ethical issues have been involved in the study.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement:Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer: Giacinto Bagetta, University of Calabria, Italy.
Additional file: Open peer review report 1.
杂志排行

中国神经再生研究(英文版)的其它文章
- Novel miRNA, miR-sc14, promotes Schwann cell proliferation and migration
- Role of behavioral training in reducing functional impairments after stroke
- Remodeling dendritic spines for treatment of traumatic brain injury
- Acute drivers of neuroinflammation in traumatic brain injury
- More than anti-malarial agents: therapeutic potential of artemisinins in neurodegeneration
- Why microglia kill neurons after neural disorders?The friendly fire hypothesis