Strategies to promote the maturation of ALS-associated SOD1 mutants:small molecules return to the fold
2019-07-17LukeMcAlary,JustinJ.Yerbury
(Cu)IIATSM (CuATSM) promotes the proper folding of familial amyotrophic lateral sclerosis (fALS)-associated copper, zinc superoxide dismutase (SOD1): ALS is a progressive neurodegenerative disease that affects motor neurons in the cortex and spinal cord, resulting in paralysis and ultimately death. The majority of cases are sporadic and the remainder are fALS where a subset of cases are associated with over 160, mostly missense, mutations in the SOD1 enzyme structure. Generally, these mutations impede the correct folding of SOD1 by disrupting either metal binding,disulfide formation, and/or dimerization, leading to the accumulation of misfolded SOD1 which can form the intracellular inclusions observed in patient tissue. The misfolding of SOD1 causes downstream effects such as ubiquitin proteasome dysfunction,endoplasmic reticulum stress, mitochondrial dysfunction, and calcium dyshomeostasis. Indeed, the thermal and chemical stability of SOD1, including fALS mutants, is significantly increased upon binding of metals and/or disulfide formation. Recently, a copper(Cu)-based small molecule called CuATSM was found to be effective at treating multiple transgenic mouse models expressing human SOD1-fALS protein (Soon et al., 2011; Roberts et al., 2014). It was found that CuATSM treatment increased the levels of soluble Cu-bound SOD1 whilst still prolonging disease progression and time until onset of symptoms. Although this was a great success,a gap in knowledge existed as to whether CuATSM would be an effective treatment in cases where the SOD1-fALS mutant has perturbed or ablated Cu-binding. We recently showed that CuATSM is not effective at rescuing the toxicity associated with the expression of SOD1 metal-binding-region mutants in cultured cells (Farrawell et al., 2018). This suggests that although CuATSM has many therapeutic benefits not associated with SOD1 maturation, it may not be as effective for patients carrying SOD1-fALS metal-binding-region mutations and, hence, other therapeutic options may need to be pursued in these cases.
SOD1 maturation and misfolding: Although it is a small protein,SOD1 undergoes extensive post-translational modification to reach its native dimeric conformation. Following synthesis, SOD1 assumes a partially folded state primed for zinc binding, which occurs spontaneously in the cell. After Zn is bound, SOD1 will associate with its co-chaperone ‘the copper chaperone for SOD1'(CCS), which delivers Cu and oxidizes the intramolecular disulfide bond. The mature monomer is then available to associate with other mature monomers to form dimers. At each step SOD1 is susceptible to proceeding down off-folding pathways, primarily due to its metastability in cells (Ciryam et al., 2017). It is established that the cytotoxicity associated with mutant SOD1 in fALS is a result of misfolding and subsequent aggregation of the protein. Interestingly, the physical properties of SOD1 mutants have been exhaustively studied, finding that different mutants of SOD1 can affect folding stability to varying degrees, providing a plausible physico-chemical rationale for the variable toxicities of different mutants. Altogether,this evidence suggests that therapeutics that can stabilise mature states of SOD1, or lower the kinetic barriers to form mature states,would be effective at alleviating pathology in SOD1-fALS.
Targeting dimerization:Some past attempts at developing SOD1-targeting pharmacological chaperones have focused around promoting the formation and maintenance of SOD1 in its dimeric state (Ray et al., 2005; Capper et al., 2018). Examples include the use of cysteine reactive crosslinking chemicals to potentially promote the formation of stable dimers through covalent linking between cysteine-111 residues (Auclair et al., 2010). Whilst effective at stabilizing SOD1 in in vitro experiments, a caveat to their use as a therapeutic is that compounds that promote covalent protein-protein crosslinks are highly toxic owing to their lack of specificity to the target protein. More recently, the selenium-based antioxidant compound ebselen was shown to significantly increase the dimer-binding affinity of SOD1-fALS mutants in vitro and exert minimal toxicity to cultured cells (Capper et al., 2018). The increase in dimer affinity was thought to arise from planar π-π interactions between ebselen molecules covalently bound to cysteine-111 through a seleno-sulhpur bond on adjacent SOD1 monomers. Non-covalent binding approaches have also been tested,where several compounds that bind within the SOD1-A4V (alanine to valine substitution at amino acid position 4) dimer cavity were found to stabilize the dimer and make the mutant more resistant to aggregation in vitro (Ray et al., 2005). Several of the identified compounds in this study contained aromatic moieties that were determined to dock into a hydrophobic pocket between adjacent SOD1 monomers. The concept of stabilizing dimers works in vitro because these simple models start with folded SOD1 and typically require induction of dimer dissociation which subsequently results in misfolding and toxic aggregation prone species. However,the strategy of stabilizing dimers may not be entirely effective for SOD1-fALS in vivo as dimerization occurs late in the maturation pathway of SOD1, making therapeutics that promote metal binding or disulfide formation more attractive as they affect SOD1 folding at an earlier stage. Regardless, these therapeutics represent an important class that warrant further investigation.
Targeting metal binding:Excitingly, CuATSM has been established to facilitate increased Cu-binding of SOD1 in cell culture models and animal models, thereby increasing the pools of stable SOD1 in the cell and increasing the barrier to misfolding of the bulk protein population (Soon et al., 2011; Roberts et al., 2014; Farrawell et al., 2018). However, as stated earlier, this increase in Cu binding seems to solely affect those mutations that do not disrupt Cu coordination (Farrawell et al., 2018). The drug appears to act through increasing the level of Cu-bound CCS, which then goes on to deliver copper to SOD1. The drug also has other noted benefits that are not associated with SOD1, evidenced by the effectiveness of the drug in improving motor function and decreasing neurological pathology in murine Parkinson's disease models (Hung et al.,2012). Currently, a phase I clinical trial is underway for CuATSM in ALS patients in Australia.
Other than Cu-binding, increasing Zn binding is a possible therapeutic target for SOD1-fALS. Owing to Zn-binding being the first stabilizing post-translational modification that occurs to SOD1 during its maturation, and that it results in significant increases in stability, it would be an obvious target for therapeutic intervention.Zn supplementation has been explored previously, resulting in increased survival of mice expressing human SOD1-G93A (Ermilova et al., 2005). There is currently no effective delivery molecule for Zn into the central nervous system and, therefore, any increase of Zn concentration in the central nervous system would have to occur through passive administration or supplementation. However, this may be problematic due to the fact that high-dose Zn supplementation interferes with the uptake of Cu.
Targeting disulfide formation:Concomitant with the transfer of Cu to SOD1 by CCS is the oxidation of the intramolecular disulfide bond between Cys57 and Cys146. This has obvious benefits to SOD1 structure by increasing the thermal stability of the protein. However, since CuATSM did not protect against the toxicity of SOD1 mutants with disrupted Cu-binding in cells, a strategy focused around the CCS-independent formation of the disulfide could prove to be effective in those cases. For example, ebselen may prove to be a useful lead compound in this class of drug, as in-cell nuclear magnetic resonance previously showed that cells expressing G93A or A4V SOD1 in the presence of ebselen were fully disulfide oxidized (Capper et al., 2018), whereas ~95% of the SOD1 expressed in the absence of ebselen existed in a disulfide reduced state. Ebselen was found to undergo a redox interaction with Znbound disulfide reduced SOD1, which facilitated the formation of the disulfide bond. Considering this, one would expect ebselen to be effective as a therapeutic for SOD1-fALS, perhaps even in the case of mutants with perturbed Cu-binding, although experiments in cell and animal models would need to be performed to further investigate ebselen as a possible small molecule therapeutic for SOD1-fALS.
Blocking oligomerization and aggregation:Even though promotion of SOD1 maturation is an increasingly attractive therapeutic avenue, blocking oligomerization and aggregation of SOD1 should still be pursued. This is owing to the relative effects that different mutations have on SOD1 stability and maturation, including truncation mutations such as G127X that do not fold, which translates to differential responses to drug treatments (Farrawell et al., 2018).Previously, a lead compound based on a pyrimidine-2,4,6-trione scaffold was found to be effective in blocking SOD1 aggregation and SOD1-induced cell death in cultured cells, as well as having promising pharmocological properties (Xia et al., 2011). More recently, other pyrimidine based compounds have been identified that prevent SOD1 aggregation in cultured cells or rescue SOD1-induced toxicity in vivo. 5-Fluorouridine and 5-Fluorouracil were found to significantly decrease the formation of SOD1 inclusions in cultured cells (Pokrishevsky et al., 2018) and protect against motor neuron axonopathy in zebrafish expressing human wild-type SOD1 (DuVal et al., 2019). The antiviral drug telbivudine was also found to protect against SOD1-induced toxicity in a zebrafish model, rescuing the axonopathy phenotype and motor functions of treated fish expressing human SOD1. The mode of action of 5-fluorouridine, 5-fluorouracil and telbivudine has suggested to be interaction with the tryptophan residue at position 32 in SOD1 which is important for aggregation. Regardless, it appears that pyrimidine-based compounds are a class of compound that is effective at preventing SOD1 aggregation and associated toxicity.
Conclusions and future directions:Investigation of therapeutics that target the structural consequences of mutation and/or misfolding of SOD1 have been ongoing since the discovery of the protein's association with ALS. Typically, studies have focused on preventing the aggregation of SOD1, however, recent work has provided not only new aggregation blocking compounds, but also compounds that facilitate the maturation of SOD1 to a more aggregation resistant state. Considering the multiple pathways through which SOD1 might misfold and aggregate, a combinatorial approach that utilizes compounds effective at metal loading, disulfide formation,and aggregation blocking, may prove more useful than any single compound (Figure 1). In summary, new small molecules on the horizon show some promise for treating sufferers of SOD1-fALS and should be pursued for translation into the clinic.
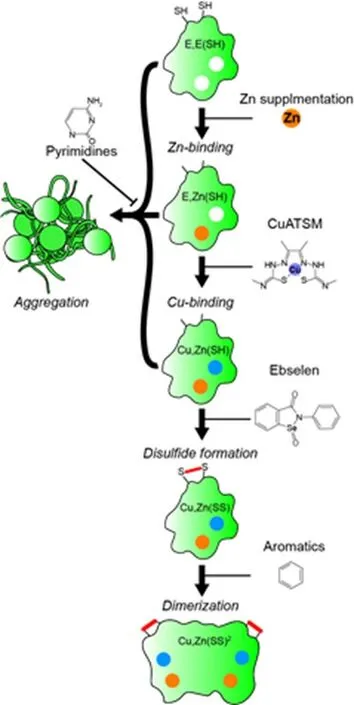
Figure 1 Strategies to prevent SOD1-associated toxicity through promotion of maturation or blockade of aggregation by small molecules.SOD1 transitions through multiple post-translational modification states prior to being in its fully mature, enzymatically active state.The diagram above shows these states with the corresponding processes that occur to obtain them(italics), as well as the possible small molecules that may be used to promote them (right). Aggregation is most likely to occur to immature SOD1 forms and can potentially be blocked by pyrimidine compounds.E,E(SH) is apo-disulfide reduced SOD1, and Cu,Zn(SS)2 is a mature dimer. SOD1: Copper,zinc superoxide dismutase.
Luke McAlary, Justin J. Yerbury*
Illawarra Health and Medical Research Institute, Wollongong,NSW, Australia (McAlary L, Yerbury JJ)School of Chemistry and Molecular Bioscience, Molecular
Horizons, Faculty of Science, Medicine and Health, University of Wollongong, NSW, Australia (McAlary L, Yerbury JJ)
*Correspondence to: Justin J. Yerbury, PhD, jyerbury@uow.edu.au.orcid: 0000-0003-2528-7039 (Justin J. Yerbury)
Received:December 27, 2018
Accepted: February 27, 2019
doi: 10.4103/1673-5374.255962
Copyright license agreement:The Copyright License Agreement has been signed by both authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer: Manoj Kumar Jaiswal, School of Medicine at Mount Sinai, USA.
Additional file: Open peer review report 1.
杂志排行

中国神经再生研究(英文版)的其它文章
- Novel miRNA, miR-sc14, promotes Schwann cell proliferation and migration
- Role of behavioral training in reducing functional impairments after stroke
- Remodeling dendritic spines for treatment of traumatic brain injury
- Acute drivers of neuroinflammation in traumatic brain injury
- More than anti-malarial agents: therapeutic potential of artemisinins in neurodegeneration
- Why microglia kill neurons after neural disorders?The friendly fire hypothesis