胃炎宁颗粒质量标准研究
2019-07-16朱家红王影超郭延红
朱家红 ,陈 贇 ,王影超,杨 勇 ,郭延红,冯 胜
(1.贵阳护理职业学院,贵州 贵阳550081;2.贵州省食品药品检验所,贵州 贵阳 550004; 3.贵阳市第四人民医院,贵州 贵阳 550002)
胃炎宁颗粒由檀香、木香(煨)、细辛、肉桂、赤小豆、鸡内金、甘草(蜜炙)、山楂、乌梅、薏苡仁(炒)十味药材组成,具有温中醒脾、和胃降逆、芳香化浊、消导化食的功效,临床用于伤食湿重引起的胃脘痛、泛酸、恶心及消化不良的治疗。目前,胃炎宁颗粒的质量控制方法为国家食品药品监督管理局颁布的WS3-B-0774-91,仅有显微鉴别及常规项目检查。为更全面地控制胃炎宁颗粒的质量,保证临床用药安全,本研究建立了该制剂中甘草和薏苡仁的薄层鉴别方法,以及甘草中甘草苷和甘草酸的含量测定方法,现报道如下。
1 仪器与试药
1.1 仪器
薄层色谱自动点样仪及照相系统(CAMAG ATS Ⅲ);高效液相色谱仪(Thermo U3000和Agilent 1260 );电子天平(METTLER TOLEDO);超纯水机(UPW-50N);KQ-500DA型数控超声波提取仪。
1.2 试剂与试药
乙腈(色谱纯,美国天地有限公司);磷酸(色谱纯,天津光复精细化工研究所,批号28080705);石油醚(分析纯,天津富宇细化工有限公司,批号20180415);甲醇(分析纯,天津富宇精细化工有限公司,批号:20180928);其余试剂均为分析纯。
甘草对照药材(120904-201620)、薏苡仁对照药材(121254-201203)、薏苡仁对照提取油(11750-201703)、甘草苷对照品(111610-201106以93.0%计)、甘草酸铵对照品(110731-201619以93.7%计)均由中国食品药品检定研究院提供。硅胶G板(安徽良臣硅源材料有限公司,批号:20170815);胃炎宁颗粒样品分别来自山东孔府制药有限公司(20180403)、湖北老中医制药有限责任公司(20180415)、长春人民药业集团有限公司(20180402)、吉林亚泰明星制药有限公司(20180413)。
2 方法与结果
2.1 薄层色谱鉴别
2.1.1 甘草薄层色谱鉴别[1-3]取样品(批号为20180415)15 g,研细,加甲醇50 mL,超声提取功率(100 kW)30 min,滤过,滤液挥干,残渣加水20 mL使其溶解,加乙醚30 mL振摇提取,弃去乙醚液,水液用水饱和的正丁醇振摇提取2次,每次30 mL,合并正丁醇液,回收溶剂至干,残渣加乙醇1 mL使其溶解,作为供试品溶液。另取甘草对照药材1 g,同法制成对照药材溶液。取缺甘草的阴性样品,按供试品溶液制备方法,制得阴性对照溶液,按照薄层色谱法(通则0502)试验,吸取上述溶液0.5~1 μL,分别点于同一硅胶G薄层板上,以乙酸乙酯-甲酸-冰醋酸-水(15∶1∶1∶2)为展开剂展开,取出,晾干,喷以10%硫酸乙醇溶液,105 ℃加热至斑点显色清晰,置于紫外光灯(365 nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光主斑点,阴性样品无干扰。见图1。

1.阴性样品;2.甘草对照药材(120904-200512);3.样品(长春人民药业集团有限公司,批号20180402);4.样品(湖北老中医制药有限责任公司,批号20180415);5.样品(吉林亚泰明星制药有限公司,批号20180413);6.样品(山东孔府制药有限公司,批号20180403)(紫外灯光365 nm下检视,温度21 ℃、相对湿度53%)
图1 甘草薄层色谱
2.1.2 薏苡仁薄层色谱鉴别[1,4-7]取样品(批号为20180415)10 g, 研细,加石油醚(60~90 ℃)50 mL,超声提取(100 kW)30 min,滤过,滤液浓缩至3 mL,作为供试品溶液,另取薏苡仁对照药材1 g,同法制成对照药材溶液。再取薏苡仁油对照提取物,加石油醚(60~90 ℃)制成1 mL含2 mg对照药材的溶液,作为对照提取物溶液。取缺薏苡仁的阴性样品,按供试品溶液制备方法,制备阴性对照溶液,按照薄层色谱法(通则0502)试验,吸取供试品溶液5 μL、对照药材和对照提取物溶液各1 μL,分别点于同一硅胶G薄层板上,以石油醚(60~90 ℃)-乙醚-冰醋酸(8∶2∶0.1)为展开溶液展开,取出,晾干,喷以10%硫酸乙醇溶液,105 ℃加热至斑点显色清晰。供试品色谱中,在与对照药材色谱和对照品提取物色谱相应的位置上,显相同颜色的斑点,阴性样品无干扰。见图2。
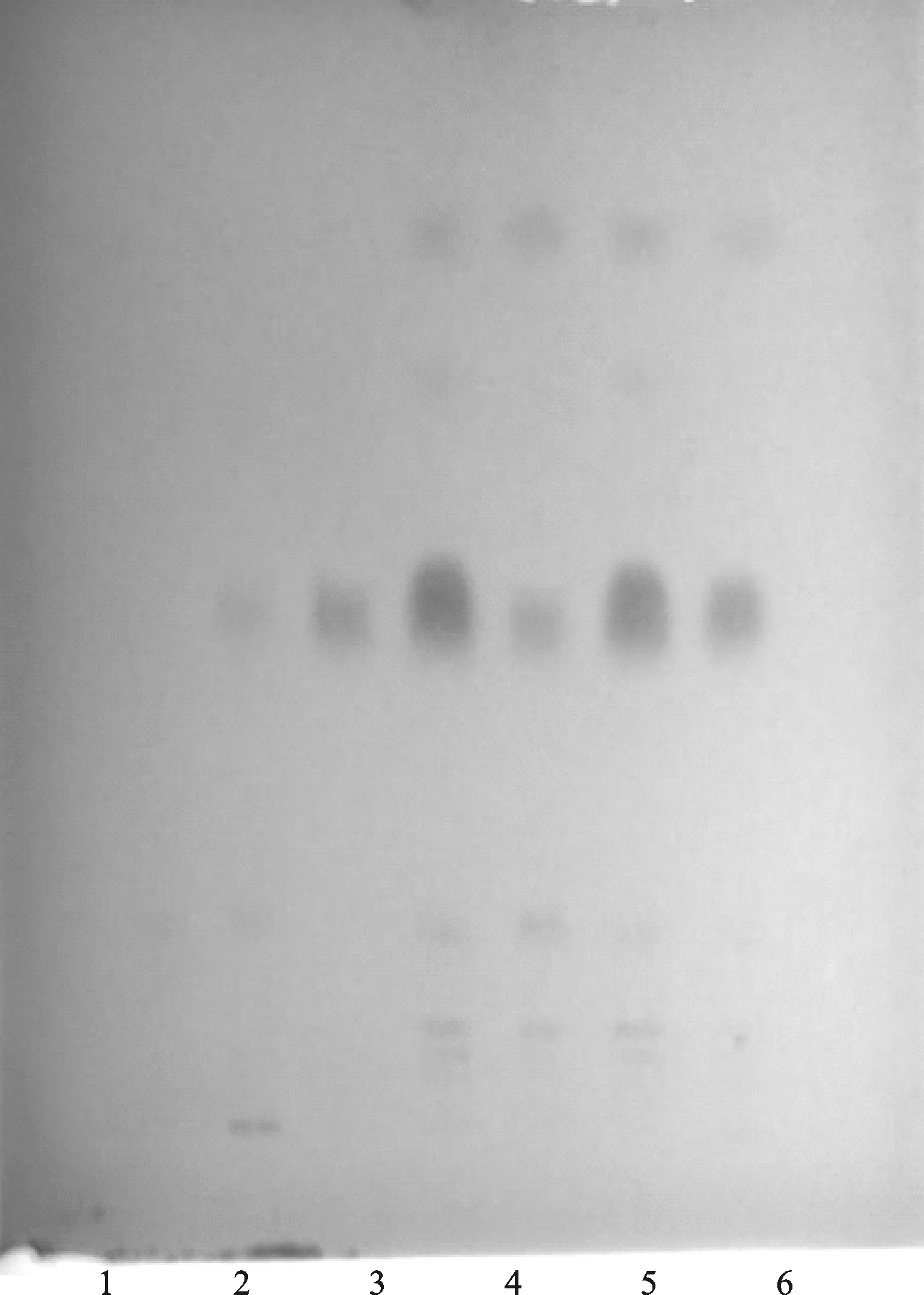
1.阴性样品;2.薏苡仁对照药材(121254-201203);3.薏苡仁对照品提取物(11750-201703);4.样品(长春人民药业集团有限公司,批号20180402);5.样品(湖北老中医制药有限责任公司,批号20180415);6.样品(吉林亚泰明星制药有限公司,批号20180413);7.样品(山东孔府制药有限公司,批号20180403)。(日光下检视,室温20 ℃,相对湿度20%)
图2 薏苡仁薄层色谱
2.2 甘草含量测定
2.2.1 色谱条件及系统适用性试验 色谱柱:Waters XBridge C18色谱柱(4.6 mn×250 mm,5 μm),流动相:乙腈-0.05%磷酸溶液,梯度洗脱程序见表1。检测波长:273 nm,流速:1.0 mL/min,柱温:30 ℃。理论塔板数按甘草苷计应不低于5 000。

表1 二元梯度洗脱程序 (%)
2.2.2 溶液制备 ①供试品溶液:取胃炎宁颗粒(批号:20180415)适量,研细,取0.25 g,精密称定,置于具塞锥形瓶中,精密加入70%乙醇25 mL,密塞,称定重量,超声提取(100 Kw)20 min,放冷,再称定重量,用70%乙醇补足减失的重量,摇匀,用0.45 μm微孔滤膜过滤,取续滤液即得;②取缺甘草药材的阴性样品,制备阴性对照品溶液;③对照品溶液:精密称定甘草苷(2.707 mg)、甘草酸铵对照品(3.493 mg),分别置于不同容量瓶中,加70%乙醇稀释至刻度,制成每1 mL含上述2种成分依次为0.253 6、0.324 8 mg的单一对照品贮备液。
2.2.3 方法学考察 (1)专属性试验。取“2.2.2”项下3种溶液,按照拟定的色谱条件进样,记录色谱峰。结果供试品溶液和对照品溶液在相同保留时间处有色谱峰,阴性样品无干扰,且分离度较好,表明方法专属性好。详见图 3。
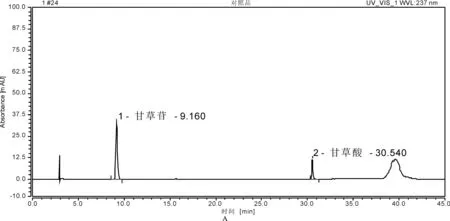
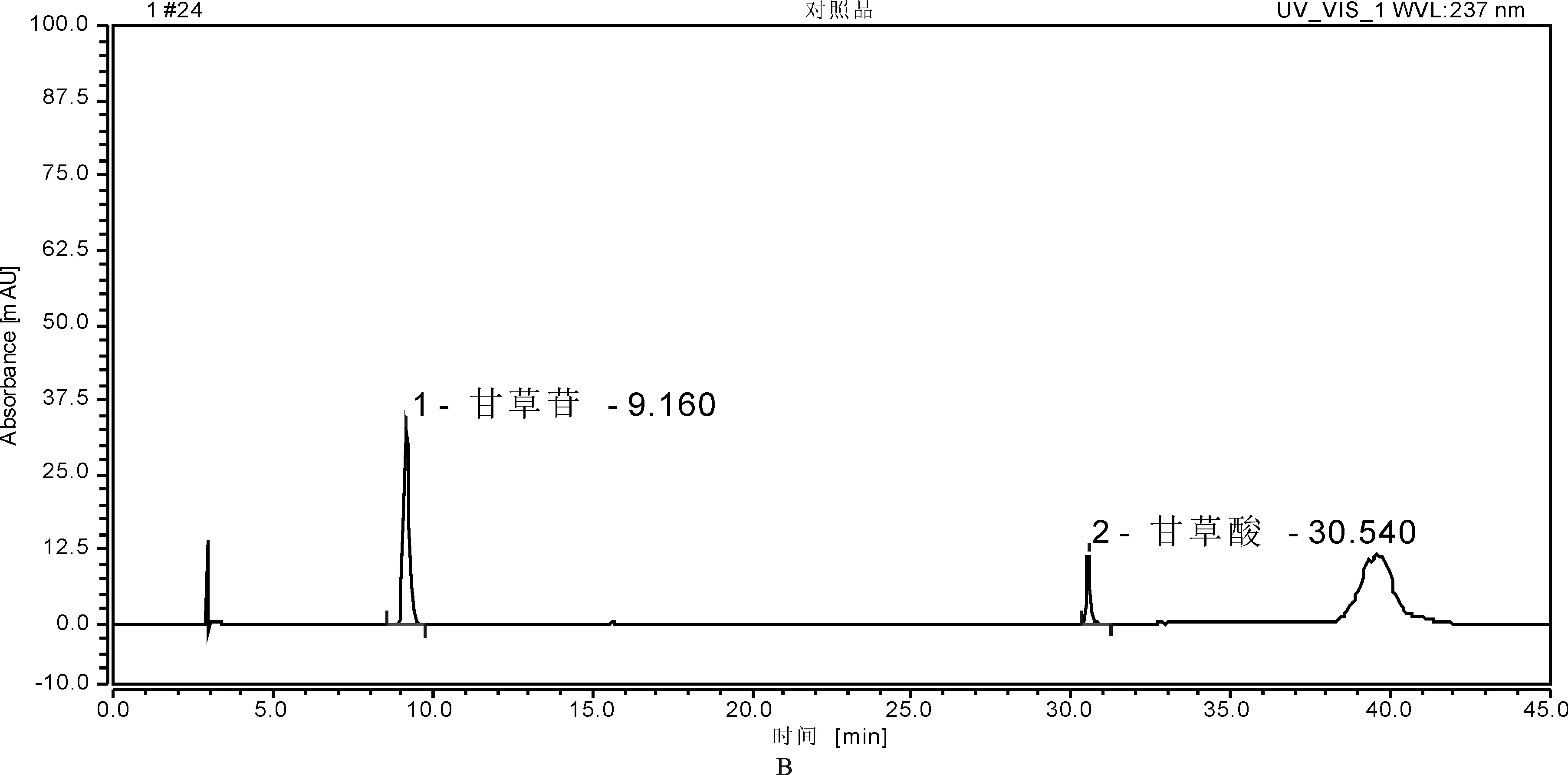
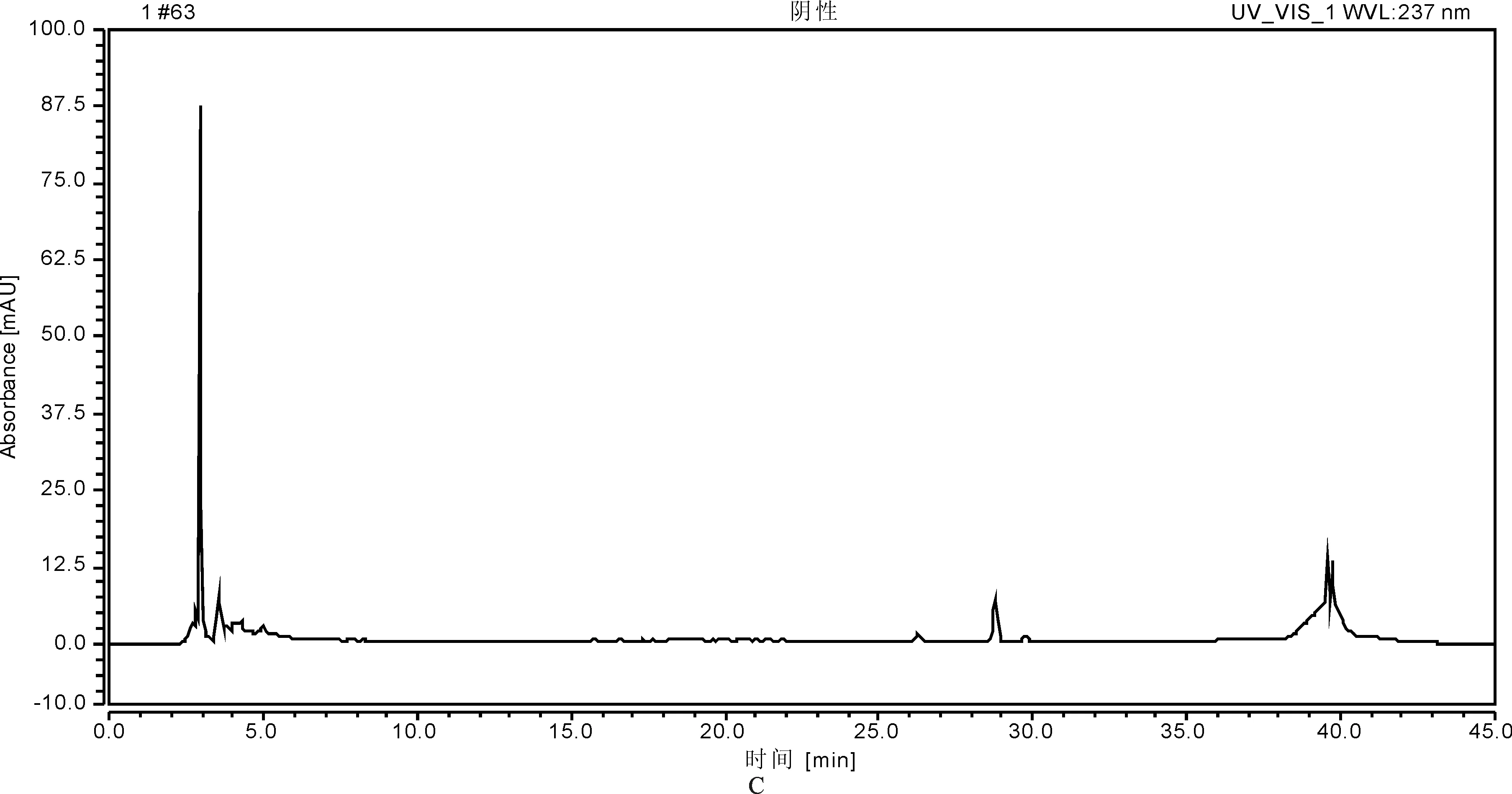
1.甘草苷;2.甘草酸 A.对照品溶液;B.样品溶液;C.阴性对照品溶液
(2)线性关系考察。精密量取“2.2.2”项下对照品贮备液各1 mL,置于同一10 mL量瓶中,加70%乙醇稀释至刻度,摇匀,依次进样1 μL、5 μL、10 μL、15 μL、20 μL;再精密量取上述对照品贮备液各1 mL,置于同一2 mL量瓶中,摇匀。 进样量10 μL,按上述色谱条件分别进样测定,以进样量(X)为横坐标,峰面积为(Y)纵坐标绘制标准曲线,计算回归方程和相关系数(见表2)。结果表明,2种成分在各自的浓度范围内与峰面积积分线性关系良好。
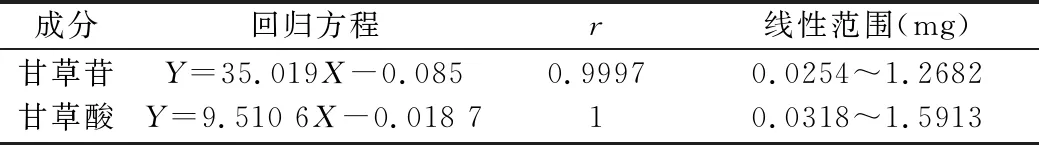
表2 两种指标成分的线性关系和范围
(3)精密度考察。精密吸取甘草苷(0.253 6 mg/mL)、甘草酸铵(0.318 3 mg/mL)的对照品混合溶液10 μL,重复进样6次,结果甘草苷峰面积RSD为0.56%(n=6),甘草酸峰面积RSD为0.11%(n=6),表明仪器精密度良好。
(4)重复性试验。取同一批(批号为20180415)样品,按照“2.2.2”项下方法制备供试品溶液6份,按拟定色谱条件测定,结果甘草苷与甘草酸的平均含量分别为0.309 5 mg/g、0.374 9 mg/g,RSD分别为1.17%(n=6)、1.06%(n=6),表明方法重复性良好。
(5)稳定性试验。取同一批供试品溶液(批号为20180415),分别于放置0、4、8、10、13、21、24 h时进样,测定甘草苷的峰面积,结果RSD为 0.669 8%(n=6);测定甘草酸的峰面积,结果RSD为0.358 2%(n=6)。表明供试品溶液在24 h内稳定。
(6)加样回收试验。取样品(批号为20180415,甘草苷含量为0.386 9 mg/g,甘草酸含量为0.468 6 mg/g)6份,精密称定,每份0.25 g,置于具塞锥形瓶中,精密加入甘草苷(0.384 076 3 mg)和甘草酸铵(0.475 95 mg)的混合对照品溶液 25 mL,按照“2.2.2”项下方法制备供试品溶液,进样测定,计算回收率。结果见表3。
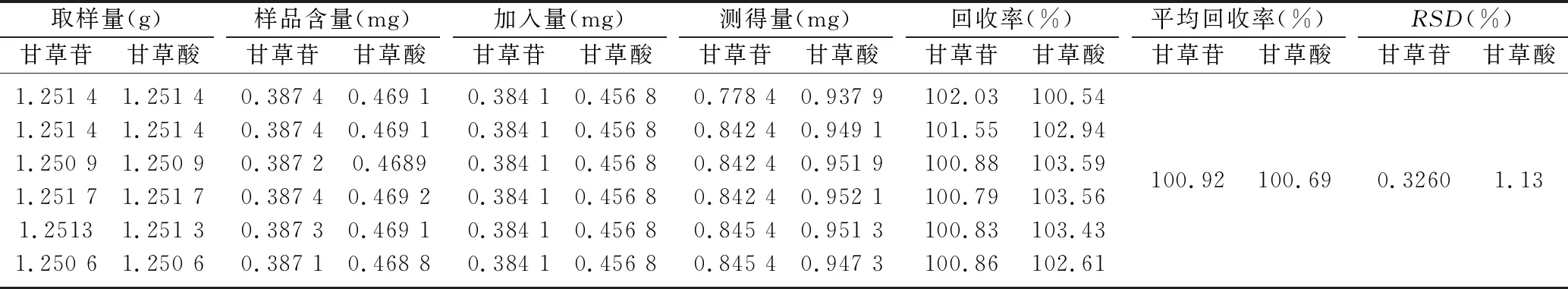
表3 甘草苷和甘草酸加样回收试验结果 (n=6)
(7)样品测定。按照“2.2.1”项下色谱条件,分别测定4批样品,结果见表4。
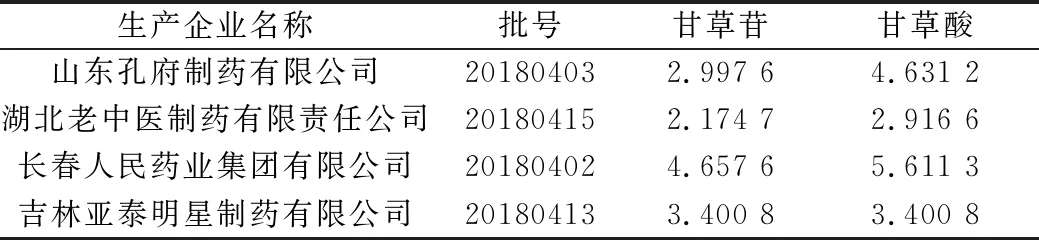
表4 不同厂家胃炎宁颗粒中甘草苷和甘草酸含量 (mg/袋)
3 讨论
3.1 定性鉴别
参考《中国药典》及有关文献,方中乌梅、山楂的主要成分均为枸橼酸[1],无法确定专属性,目前国内也没有相应研究,故不选择这两种进行研究。根据工艺方法得知檀香、木香(煨)细辛、肉桂的有效成分均为挥发油,曾测得木香的主要成分木香烃内酯和去氢木香烃内酯含量太低,考察认为在方中挥发油药材占比低,且在生产中损失比较大,故不选择木香作为研究对象。
甘草薄层板:考察了2015版《中国药典》甘草的薄层板,即1%氢氧化钠的硅胶G板,分离效果不好,故改用硅胶G板,分离效果好,斑点清晰。
薏苡仁展开剂选择:以石油醚(60~90 ℃)-乙酸乙酯-醋酸(10∶3∶0.1)为展开剂展开,Rf值小,分离效果不好,故选择石油醚(60~90 ℃)-乙醚-冰醋酸(10∶2∶0.1)为展开剂,阴性样品无干扰,分离效果好。
薏苡仁的观察条件选择:参考2015版《中国药典》薏苡仁的薄层鉴别,喷以5%香草醛硫酸溶液,在105 ℃加热至斑点清晰,使用以后斑点不明显,故选择10%硫酸乙醇溶液,在105 ℃加热,置于紫外灯(365 nm)下检视,斑点清晰,分离度好,无干扰。
3.2 含量测定
提取溶剂与方法:根据甘草苷、甘草酸的理化特性,选择甲醇、乙醇、70%乙醇为提取溶剂,超声提取,以70%乙醇提取效率最高,杂质对主峰无干扰,峰型对称,故本实验选择70%乙醇为提取溶剂。再对70%乙醇超声提取和回流提取的效率进行考察,结果显示,两种提取方式效率无明显差异,且超声提取操作简单,故最终选择超声提取。
提取温度与时间:考察了提取时间对提取效率的影响,结果显示,提取时间超过 20 min时提取效率变化不明显,故选择超声 20 min 提取。
流动相及检测波长:曾参考文献[2]采用乙腈-0.017 mol/L磷酸水溶液-三乙胺( 35∶65∶0.3) 作为流动相测定甘草酸铵( 实际测得峰是甘草酸的峰,再按系数折算成甘草酸铵,即甘草酸铵的量=甘草酸的量×1.020 7) ,结果未出峰,原因是流动相偏碱性,甘草酸铵无法水解成甘草酸,故最终选择乙腈(A)-0.05%磷酸水溶液(B)为流动相,梯度洗脱(0 min时82%B,8 min时82%B,35 min时50%B,36 min时0%B,45 min时82%B )。本试验中选择 273 nm 为甘草苷与甘草酸铵的检测波长。
