Mutant p53 increases exosome-mediated transfer of miR-21-3p and miR-769-3p to promote pulmonary metastasis
2019-07-13QiangJuLinaZhaoJiajiaGaoLanpingZhouYangXuYulinSunXiaohangZhao
Qiang Ju, Lina Zhao, Jiajia Gao, Lanping Zhou, Yang Xu, Yulin Sun, Xiaohang Zhao
State Key Laboratory of Molecular Oncology, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100021, China
Abstract Objective: Tumor metastasis is a complex, multistep process that depends on tumor cells and their communication with the tumor microenvironment. A p53 gain-of-function mutant has been shown to enhance the tumorigenesis, invasion, and metastasis abilities of tumor cells. This study aimed to investigate the roles of p53 R273H mutation in the tumor microenvironment.Methods: The in vitro and in vivo effects of the p53 R273H mutant on the invasion and metastasis of HCT116 cells were investigated. Exosomes from wild-type and HCT116-TP53(R273H) cells were cocultured with mouse embryonic fibroblasts (MEFs). The roles of differentially expressed exosomal microRNAs identified by microarray analysis were investigated. The functions of the p53 R273H mutant in tumor cells were also investigated via gene expression microarray and quantitative polymerase chain reaction (qPCR) analyses.Results: Introducing p53 R273H mutant into HCT116 cells significantly potentiated pulmonary metastasis in vivo. In the presence of exosomes derived from HCT116-TP53(R273H) cells, the exosomes were taken up by MEFs and became activated. Microarray analysis showed that the p53 R273H mutation increased the exosomal levels of miR-21-3p and miR-769-3p. Intriguingly, in clinical samples, miR-21-3p and miR-769-3p levels were significantly higher in patients with a p53 mutation than in those without this mutation. Furthermore, both miR-21-3p and miR-769-3p activated fibroblasts and exerted a synergistic effect via their target genes on the transforming growth factor-β (TGF-β)/Smad signaling pathway. The activated fibroblasts excreted cytokine TGF-β and may have reciprocally induced cancer cells to undergo epithelial-mesenchymal transition (EMT). Indeed, HCT116-TP53(R273H) cells showed increased expression of ZEB1 and SNAI2 and decreased transcription of several cell adhesion molecules.Conclusions: The mutant p53-exosomal miR-21-3p/miR-769-3p-fibroblast-cytokine circuit appears to be responsible for communication between tumor and stromal cells, with exosomal miRNAs acting as a bridge. miR-21-3p and miR-769-3p are potential predictive markers of pulmonary metastasis and candidate targets for therapeutic interventions.
Keywords: miR-21-3p; miR-769-3p; exosome; tumor metastasis; tumor microenvironment
Introduction
Metastasis is the end stage of tumor progression, and the resulting metastatic diseases remain largely incurable and account for more than 90% of cancer-associated mortality(1). While the metastatic program mechanisms are not yet well understood, the intrinsic phenotypic features of tumor cells and their interactions with the tumor microenvironment and immune system are known to play pivotal roles.
In tumor cells themselves, numerous genetic and epigenetic regulators have been found to contribute to metastasis (2). Additionally, it is now accepted that acquisition of an epithelial-mesenchymal transition (EMT)phenotype strongly enhances the invasion and dissemination capabilities of tumor cells. Moreover, EMT allows cells to gain the cancer stem cell phenotypes that facilitate survival (3), and the tumor microenvironment plays critical roles in the multistep metastasis cascade. In primary tumors, the tumor microenvironment promotes tumor proliferation, invasion, survival, and angiogenesis and induces tumor cell EMT. In the distant organ, the stromal microenvironment generates a specialized premetastatic niche to foster the survival and expansion of metastatic lesions (2,4). Recent studies indicate that exosomes, a group of lipid bilayer vesicles that contain DNA, RNA, and proteins ranging in size from 30 to 100 nm, play a key role in the formation of this niche.Exosomes can be formed and secreted by primary tumor cells and alter the homeostasis of the tumor microenvironment either by directly targeting stromal cells and immune cells or by altering the structure and composition of the extracellular matrix (5,6). Moreover,exosomes can potentially home to distant organs, facilitate premetastatic niche formation and determine the metastatic organ tropism (7,8).
p53 is one of the most intriguing master regulators of metastasis (9). For example, p53 suppresses EMT and EMT-associated stem cell properties through the miR-34a/b/c-mediated downregulation of Snail, MDM2-mediated degradation of Slug, and transcriptional activation of miR-200c (10-12). Notably, in more than 50% of human tumors, p53 contains missense mutations in exons 4-9 that disrupt its DNA-binding ability. Among the common hotspot mutations in p53, introduction of the R273H (R270H in mice) and R175H (R172H in mice)mutations was shown to markedly increase the incidence of highly metastatic carcinomas in various mouse models(13,14). Subsequent evidence showed that mutant p53 promotes EMT by interfering with primary miRNA processing, upregulating Twist1, REGγ and EZH2,antagonizing KLF17 and modulating the miR-130b-ZEB1 axis (15-19).
Nevertheless, the role of p53 in the tumor microenvironment is still poorly understood. Most recently,mutant p53 was found to facilitate a chronic proinflammatory tumor microenvironment by regulating secreted interleukin-1 receptor antagonist (sIL-1Ra) (20).Additionally, mutant p53 activates fibroblasts through the secretion of inflammatory cytokines and chemokines and activates macrophages through exosomal miR-1246 (21,22).However, as pivotal mediators of premetastatic niche formation, the roles of exosomes in metastasis resulting from p53 mutation remain unclear. Our previous study showed that exosomes of p53 R273H mutant HCT116 cells are smaller and exhibit altered protein contents compared with those of p53 wild-type cells (23). In this study, we investigated the effect of the p53 R273H mutant on the metastasis ability of HCT116 cells. Exosomes from p53 mutant cells were demonstrated to induce fibroblast activation and then promote HCT116 cell metastasis to the lungs. Thus, this study might expand our understanding of p53 gain-of-function mutations in the tumor microenvironment.
Materials and methods
Reagents
Anti-p53 antibodies (cat. no. sc-126) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-α-SMA (cat. no. 14968), anti-vimentin (cat. no. 5741), anti-Smad2/3 (cat. no. 8685s), anti-p-Smad2/3 (cat. no. 8828),and anti-Skp1 (cat. no. 2156) antibodies were purchased from Cell Signaling Technology (Boston, MA, USA). Antiβ-actin (cat. no. A5316) antibodies and 6-diamidino-2-phenylindole (DAPI) were obtained from Sigma-Aldrich(St. Louis, MO, USA).
Cell lines and culture
The human colorectal cancer cell line HCT116-TP53(WT)was purchased from American Type Culture Collection(Rockville, MD, USA), and the HCT116-TP53(R273H)cell line was established by the State Key Laboratory of Molecular Oncology, Cancer Hospital of Chinese Academy of Medical Science (23). Mouse embryonic fibroblast(MEF) cells were kindly gifted by Dr. Hongbin Zhang at Peking Union Medical College. All cell lines were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin at 37 °C in 5% CO2.
Cell invasion assay
Cell invasion was measured using Transwell chambers coated with Matrigel (Corning Inc., NY, USA) as described previously (24).
Wound healing assay
Cells were seeded in a 6-well plate at a density of 7×105cells/well and cultured until confluence. A white pipette tip was used to introduce a straight scratch simulating a wound, and the medium was then replaced with serum-free RPMI 1640 medium. Images were taken at 0, 12 and 24 h after scratching using a 10× objective.
Cell adhesion assay
A 96-well plate was coated with 20 μg/mL human fibronectin (Millipore, Temecula, CA, USA) for 1 h at room temperature and then blocked with 2% bovine serum albumin (BSA) overnight at 4 °C. The cells were seeded at a density of 4×104cells/well and cultured for 15, 30, and 60 min. Cell counting kit 8 (CCK8) reagent (10 μL) was added to each well and allowed to react for 2 h at 37 °C,after which the absorbance was detected at 450 nm in a Synergy H1 Multi-Mode Reader (Bio-Rad). The other cell cultures were then rinsed 3 times with 10% formalin and stained with crystal violet for 5 min at room temperature.Images were subsequently obtained using a 10× objective.
Western blot analysis
Protein was extracted from HCT116-TP53(WT) cells,HCT116-TP53(R273H) cells, or MEFs transfected with a miRNA mimic. Western blot analysis was performed as previously described (25).
RNA extraction
Total RNA was extracted from the HCT116-TP53(WT)and HCT116-TP53(R273H) cell lines using the mirVanaTMRNA Isolation Kit (AM1561). RNA was recovered from the exosomal samples with the Total Exosome RNA and Protein Isolation Kit (Thermo Scientific, Waltham, MA, USA) according to the manufacturer’s protocol. Total RNA was quantified using the NanoDrop ND-2000 spectrophotometer (Thermo Scientific), and RNA integrity was assessed using the Agilent Bioanalyzer 2100 (Agilent Technologies).
Isolation, identification and labeling of exosomes
Exosomes were purified from conditioned media using ExoQuickTMExosome Precipitation Solution (EXOQ20A-1, SBI) and identified by electron microscopy as described previously (26). The exosome size distribution was analyzed using the NanoSight NS500 platform according to the manufacturer’s protocols (Malvern Instruments, Malvern,UK) (27). Exosomes [phosphate buffer solution (PBS) as a control] were labeled by BODIPY-ceramide (D7540,Invitrogen) as previously described (28).
Exosome internalization and fibroblast activation assay
Labeled exosomes were quantified by staining with Coomassie Brilliant Blue G-250 and diluted to 20 μg/mL in growth medium. Before the uptake assay, MEF cells were placed on chamber slides and incubated for 24 h. The uptake assay was performed as previously described (29). As a control, MEF cells were treated with PBS. The cells were observed under a fluorescence microscope. For the fibroblast activation assay, the expression of markers for fibroblast activation was detected by Western blot and quantitative real-time polymerase chain reaction (qRTPCR) analyses.
Profiling of exosomal miRNA
Sample labeling, Agilent Human miRNA microarray(8×60K, design ID: 070156) hybridization, and washing were performed based on the manufacturer’s standard protocols. Differentially expressed miRNAs were then identified on the basis of fold changes, and P values were calculated using the t-test. The thresholds set for upregulated and downregulated genes were a fold change≥2.0 and a P value ≤0.05. The target genes of differentially expressed miRNAs were predicted from the intersection of the results of 3 databases (TargetScan, microRNA.org,PITA). Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses were applied to determine the roles of these target genes.
Gene expression analysis
Sample labeling, Affymetrix Human HTA2.0 GeneChip hybridization, and washing were performed based on the manufacturer’s standard protocols. The thresholds set for upregulated and downregulated genes were a fold change≥2.0 and a P value ≤0.05. Subsequently, GO and KEGG analyses were applied to determine the roles of these differentially expressed mRNAs in association with GO terms or pathways.
qRT-PCR
Total RNA containing miRNA was reverse transcribed into cDNA using the miRcute miRNA First-Strand cDNA Synthesis Kit (TIANGEN, China) according to the manufacturer’s protocol. PCR was performed using the miRcute miRNA qPCR Detection Kit (SYBR Green)(TIANGEN) with the manufacturer-provided reverse primer and miRNA-specific forward primers (cat. nos.:CD201-0340, CD201-0336, CD201-0453, and CD201-0093, Qiagen). MiR-30a-5p and miR-30e-5p were used as endogenous controls to normalize exosomal miRNA expression (30).
For the detection of target genes, qRT-PCR was performed as previously described (31). The primers for the target genes are listed in Supplementary Table S1.
Transfection
The MEF cell line was transfected with a miRNA mimic or mimic control (Ribobio, Guangzhou, China) at 80 nmol/L using Lipofectamine 3000 according to the manufacturer’s protocol (Invitrogen, USA). After transfection for 48-96 h,the cells were used in subsequent experiments.
Animal experiments and histological assessments
A total of 5×106HCT116-TP53(WT) or HCT116-TP53(R273H) cells were suspended in 100 μL of PBS and injected into the enterocoelia of female nude mice (n=10).After 1 month, each mouse was euthanized. The resulting tumor weights were measured using an electronic balance,and the tumors, lungs, and liver were fixed in 4% neutral buffered formaldehyde and processed, embedded, and sectioned for histological assessment. All experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at the Cancer Hospital of Chinese Academy of Medical Science.
For histological assessment, the samples were either stained by hematoxylin and eosin (H&E) to reveal the degree of pathological differentiation and metastatic lesions or analyzed by immunohistochemistry (IHC) using antibodies against E-cadherin, vimentin, and α-SMA as previously described (31). Images were obtained under a Leica DMI3000 B phase-contrast microscope.
Data mining and statistical analysis
Publicly available miRNA, mRNA and exome sequencing data of 425 colon cancer patients from The Cancer Genome Atlas (TCGA) were downloaded from the NCI Genomic Data Commons (GDC; https://gdc.cancer.gov/)and cBioportal (http://www.cbioportal.org) databases. The correlations between different genes were calculated by the Spearman’s rank correlation test. Mann-Whitney U tests were used to compare the reads per million (RPM) between two groups.
Results
p53 R273H mutation promotes HCT116 cell metastasis in vitro and in vivo
To study the function of the p53 R273H mutant in the occurrence and development of colorectal carcinoma, our group previously established the HCT116-TP53(R273H)cell line (23). Sanger sequencing indicated that codon 273 of TP53 expressed in HCT116-TP53(R273H) cells was mutated from wild-type arginine (CGT) codon to histidine(CAT) (Supplementary Figure S1A). This change leads to increased p53 expression in HCT116-TP53(R273H) cells(Supplementary Figure S1B). Importantly, introduction of TP53(R273H) mutation promoted cell migration and invasion while reducing cellular adhesion ability(Figures 1A-C).
Moreover, HCT116-TP53(R273H) cells were intraperitoneally injected into mice to study the effect of p53 R273H mutation in vivo. Intriguingly, the TP53(R273H) mutation had a slight effect on the tumorigenicity of HCT116 cells in vivo (Figure 1D).However, H&E staining of tumor tissues, lungs and livers showed that p53 R273H mutation did not promote liver metastasis, but pulmonary metastases were remarkably numerous and large in the HCT116-TP53(R273H) group(Figure 1E). Therefore, these results showed that p53 R273H mutation promoted pulmonary metastasis of HCT116 cells due to their high invasion ability.
p53 R273H mutation activates fibroblasts in tumor microenvironment through exosomes
Fibroblasts are the major stromal cells in the lung, and increasing evidence has indicated that exosomes from tumor cells can induce the differentiation of fibroblasts into myofibroblast phenotypes to induce a premetastatic niche(8,32). Therefore, we hypothesized that p53 R273H mutation promotes pulmonary metastasis of HCT116 cells by activating fibroblasts.
First, we treated MEFs with conditioned medium collected from HCT116-TP53(WT) and HCT116-TP53(R273H) cells for 3-5 d. The conditioned medium from p53 mutant cells significantly induced the expression of α-SMA and vimentin in MEFs compared with that achieved with wild-type cell medium (Figure 2A,B),indicating that fibroblasts were activated by the mutant cells. Subsequently, exosomes were extracted and identified using electron microscopy and nanoparticle tracking analysis (NTA) by NanoSight (Supplementary Figure S1C,D). Thereafter, MEFs were cocultured with exosomes secreted by HCT116-TP53(R273H) cells. Similar results were observed at both the protein and transcriptional levels in MEFs treated with conditioned medium (Figure 2C,D).In addition, the mRNA expression of TGF-β2/3 in fibroblasts was dramatically increased when MEFs were treated with exosomes isolated from p53 mutant cells(Figure 2D).
To further confirm that fibroblast activation was induced by exosomes, exosomes were labeled with BODIPY ceramide and cocultured with fibroblasts for 12 h, revealing that MEFs could absorb exosomes (Figure 2E). Therefore,p53 R273H mutant cells activated fibroblasts in tumor microenvironment through exosomes.
MicroRNA expression profiles of exosomes from HCT116-TP53(WT) and HCT116-TP53(R273H) cells
Agilent bioanalyzer profiles showed that 18S and 28S ribosomal RNAs were not among the small RNAs found within exosomes, and p53 R273H mutation increased exosomal small RNA contents (Supplementary Figure S1E).Agilent human miRNA chip analyses illustrated that p53 R273H mutation induced 139 differentially expressed exosomal miRNAs, which included 104 upregulated and 35 downregulated miRNAs (Supplementary Table S2,Figure 2F).
Target gene prediction for the differentially expressed miRNAs was conducted using the TargetScan, PITA, and microRNA.org databases, and the integrated gene list was then subjected to GO and KEGG analyses. The elucidated biological functions of target genes were mainly related to transcriptional regulation and signal transduction, and these genes mainly participated in tumor signaling pathways, MAPK signaling pathways, actin cytoskeleton regulation, focal adhesion, and Wnt signaling pathways(Supplementary Figure S1F); moreover, cytoskeleton regulation, focal adhesion and Wnt signaling pathways might be associated with fibroblast activation.Exosomal miR-21-3p and miR-769-3p activate fibroblasts
miR-21-3p and miR-769-3p were further subjected to validation by qRT-PCR, revealing that p53 R273H mutant increased exosomal miR-21-3p and miR-769-3p levels(Figure 3A), which was in accordance with the differential miRNA expression profile in exosomes. After transfection of an 80 nmol/L of miR-21-3p mimic, the protein and mRNA expression levels of α-SMA, vimentin, and TGF-βs were increased in MEFs (Figure 3B,C), indicating that miR-21-3p in exosomes activated fibroblasts. Similarly, after 2 d of 80 nmol/L of miR-769-3p mimic treatment, the transcription levels of α-SMA, vimentin, and TGF-βs were increased. After 3 d, the protein expression of α-SMA was significantly increased (Figure 3D,E), indicating that miR-769-3p also activated MEFs.
Target gene prediction revealed Smad7 and Skp1 as potential target genes of miR-21-3p and miR-769-3p,respectively. Smad7 and Skp1 act on the TGF-β/Smad signaling pathway, and Skp1 specifically targets Smad3. To further validate these findings, MEFs were transfected with a miR-21-3p or miR-769-3p mimic. The expression of Smad7 and Skp1 was significantly decreased, while the expression of α-SMA, Smad2/3, and p-Smad2/3 was increased (Figure 3B-D), indicating that Smad7 and Skp1 are potential targets and that both miRNAs activate fibroblasts through the TGF-β/Smad signaling pathway.
Cotransfection of miR-21-3p and miR-769-3p mimics showed that miR-21-3p and miR-769-3p had synergistic effects on MEF activation (Figure 3F,G). In addition, after cotransfection, the protein levels of Smad2/3 and p-Smad2/3 were significantly increased compared to those after the treatment of MEFs with miR-21-3p or miR-769-3p alone (Figure 3F), indicating that the synergistic effect occurred through the Smad signaling pathway. Therefore,miR-21-3p and miR-769-3p in exosomes might synergistically activate fibroblasts through the Smad signaling pathway.
Activated fibroblasts might be responsible for pulmonary metastasis of HCT116-TP53(R273H) cells in vivo
Activated fibroblasts secrete TGF-βs, which can induce tumor cells to undergo EMT and promote their invasion and metastasis (33). The IHC results showed that Ecadherin expression was decreased and vimentin expression was increased in xenograft tumor tissues (Figure 4A), which was consistent with the occurrence of EMT in tumor cells.
In addition, because fibroblasts are the major cell population in lung tissues (8), we hypothesized that exosomes from HCT116-TP53(R273H) cells might activate fibroblasts in lung tissues to form premetastatic lesions and promote pulmonary metastasis. IHC analysis of α-SMA and vimentin, two markers of fibroblast activation,showed that their levels were increased in pulmonary metastatic tissues (Figure 4B), reflecting the activation of fibroblasts in metastatic lesions. The above results indicated that activated fibroblasts might induce tumor cells to undergo EMT, form premetastatic lesions in lung tissues and promote pulmonary metastasis through circulating exosomes.

Figure 2 Exosomes derived from HCT116-TP53(R273H) cells activate fibroblasts. (A) Western blot analysis of α-SMA and vimentin in mouse embryonic fibroblast (MEF) cells cocultured with conditioned medium derived from HCT116-TP53(R273H) and wild-type cells.GAPDH was used as a loading control; (B) Relative densitometry analysis of α-SMA expression normalized to that of GAPDH revealed a significant increase in protein levels after treatment with conditioned medium from HCT116-TP53(R273H) cells; (C) Western blot analysis of MEF cells cocultured with exosomes isolated from HCT116-TP53(R273H) cells; (D) Relative gene expression of ACTA2, VIM and TGFB2/3, encoding α-SMA, vimentin and TGF-β2/3, respectively, by quantitative real-time polymerase chain reaction (qRT-PCR) in MEFs cocultured with exosomes derived from HCT116-TP53(R273H) cells for 1, 2, or 3 d. For B and D, triplicate biological repeats are displayed as WT, HCT116-TP53(WT); MT, HCT116-TP53(R273H); *, P<0.05; **, P<0.01; ***, P<0.001; (E) Fluorescence microscopy images of MEF cells treated with BODIPY-ceramide-labeled exosomes derived from the HCT116-TP53(R273H) cell line.Scale bars represent 20 μm; (F) Heat map of overlapping exosomal miRNAs; arrows indicate miR-769-3p and miR-21-3p. Red/green indicates an increase/decrease in gene expression. WT-Exo-1/2 and MT-Exo-1/2, duplicate biological repeats of exosomes extracted from HCT116-TP53(WT) and HCT116-TP53(R273H) cells, respectively.
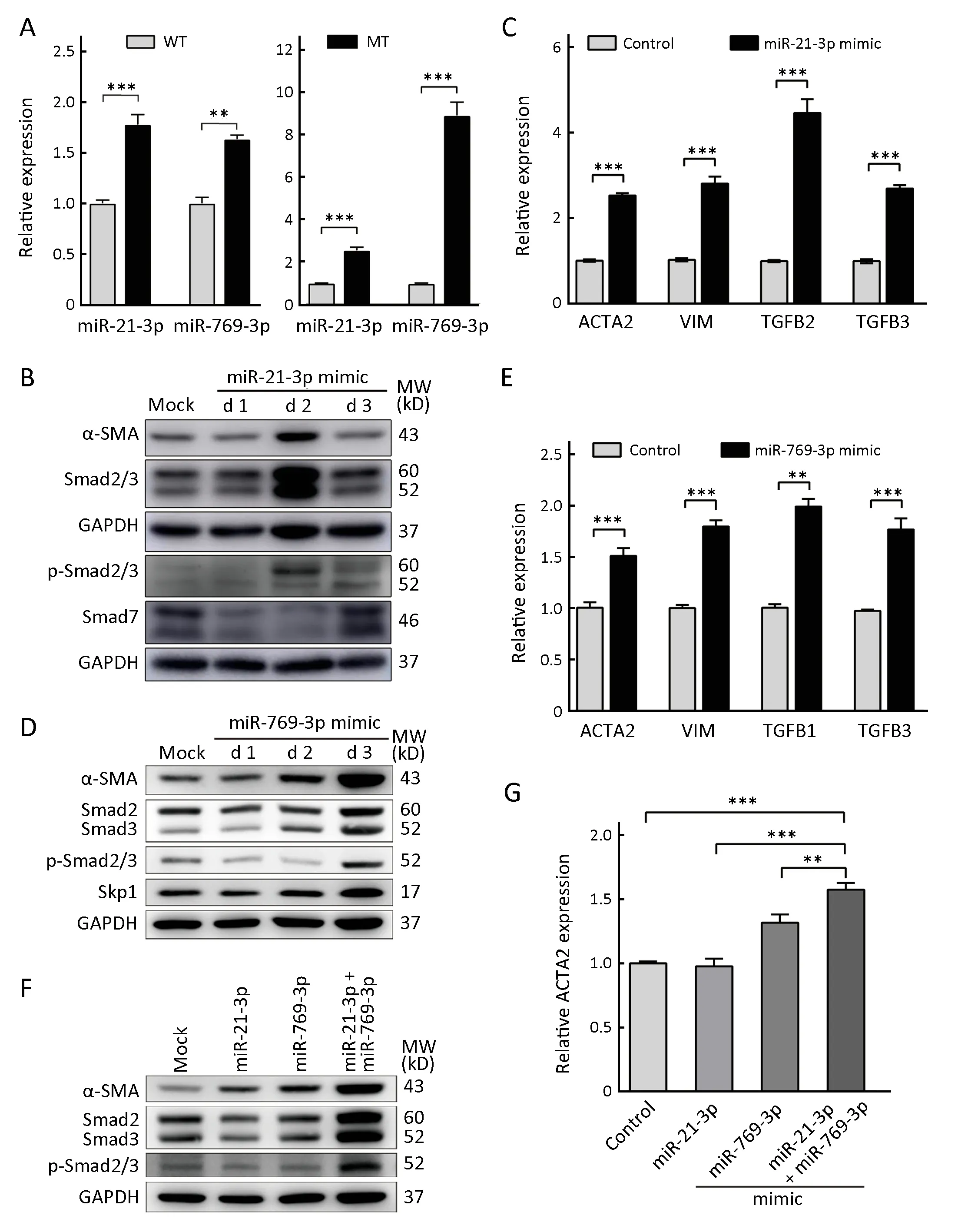
Figure 3 miR-21-3p and miR-769-3p can activate fibroblasts. (A) Intracellular (left panel) and exosomal (right panel) miR-21-3p and miR-769-3p levels were quantified by quantitative real-time polymerase chain reaction (qRT-PCR). Data representof relative miRNA levels; (B, D, F) Western blot analysis of mouse embryonic fibroblast (MEF) cells transfected with miR-21-3p, miR-769-3p or miR-21-3p combined with miR-769-3p mimics for 1, 2, and 3 d (for F, only the results after 3 d are shown). GAPDH was used as a loading control; (C,E) Relative gene expression of ACTA2, VIM and TGFB1/2/3, encoding α-SMA, vimentin and TGF-β1/2/3, respectively, by qRT-PCR in MEFs transfected with miR-21-3p (C) and miR-769-3p (E) for 2 d; (G) Relative gene expression of ACTA2, encoding α-SMA, by qRT-PCR in MEFs transfected with miR-21-3p alone, miR-769-3p alone, or miR-21-3p combined with miR-769-3p mimics for 3 d. Three biological replicates are analyzed, and the results are displayed as thep-Smad2/3, phospho-Smad2/3; mock, negative control; WT, HCT116-TP53(WT); MT, HCT116-TP53(R273H); **, P<0.01; ***, P<0.001.

Figure 4 Immunohistochemical staining of xenograft tumor tissues and pulmonary metastatic lesions. Xenograft tissues (×100)(A) and pulmonary metastatic lesions (×400) (B) were subjected to immunohistochemistry analysis of E-cadherin, vimentin, and α-SMA. WT, HCT116-TP53(WT); MT, HCT116-TP53(R273H);HE, hematoxylin and eosin.
p53 R273H mutation activates ZEB1 pathway in HCT116 cells
To clarify the effect of mutant p53 and fibroblast activation on cell phenotypes, we compared the transcriptomes of HCT116-TP53(WT) and HCT116-TP53(R273H) cells.We identified 589 differentially expressed genes exhibiting fold changes ≥2 and P values ≤0.05 in HCT116-TP53(R273H) cells, including 224 upregulated genes and 365 downregulated genes (Supplementary Table S3,Figure 5A). Pathway analysis showed that functions of downregulated genes were mainly concentrated in pathways involving cell adhesion molecules and extracellular cell matrix receptor interaction, reflecting correlation with EMT of tumor cells (Figure 5B). Thus, to further confirm the differential expression profile, we adopted qRT-PCR to validate the selected EMTassociated genes and cell adhesion molecules in the transcriptome. Consistent with transcriptome analysis, the expression of ZEB1 was increased, while the expression of GRHL2, ESRP1, MAL2, CLDN4, CLDN7, CDH3, ALCAM and OCLN was decreased in HCT116-TP53(R273H) cells(Figure 5C). Next, Western blot and qRT-PCR analyses showed that p53 R273H mutation reduced E-cadherin(CDH1) expression while increasing SNAI2 (slug) and RhoA expression (Figure 5D,E). Collectively, the results showed that p53 R273H mutation increased expression of EMT-associated transcription factor SNAI2 and decreased that of cell adhesion molecules.
MiR-21-3p and miR-769-3p expression is associated with p53 status in human colon cancer tissues
To determine whether the levels of miR-21-3p and miR-769-3p were affected by the p53 mutant in clinical human samples, we analyzed miRNA and exome sequencing data from colon cancer patients (n=425) from TCGA. We initially observed that the abundance of miR-21-3p was slightly correlated with that of miR-769-3p (rho=0.12,P=0.0102; Figure 6A). Importantly, compared with those in patients without driver mutations in TP53 gene, those with mutant p53 showed significantly higher levels of miR-21-3p and miR-769-3p in tumor tissues (Figure 6B), suggesting that the elevated exosomal miR-21-3p and miR-769-3p levels in patients with p53 mutations might result from the corresponding malignant cells. These results were consistent with our findings in the cell model.Furthermore, the expression of miR-21-3p and miR-769-3p was inversely correlated with mRNA levels of their relevant target genes Smad7 and Skp1 in tumor tissues(Figure 6C,D). Taken together, these results indicated that miR-21-3p and miR-769-3p in the exosomes of patients harboring a p53 mutation might indeed participate in metastasis via the Smad signaling pathway in clinical samples.
Discussion
In recent decades, the roles of wild-type and mutant p53 in cell migration, invasion and metastasis have been explored in various tumors. However, most of these studies have focused on the influence of p53 or its gain-of-function in tumor cells themselves. Until recently, only a few studies have described the functions of wild-type and mutant p53 in the formation of inflammatory tumor microenvironment(21,22,34). Nevertheless, it remains unclear whether there are other possible mechanisms underlying mutant p53-induced tumor microenvironment remodeling and premetastatic niche formation. In the present study, we concentrated on exosomes, which are important mediators of tumor-host cell communication. We found that TP53 driver mutations in colorectal carcinoma promoted in vivo pulmonary metastasis and altered the composition of exosomal miRNAs. Notably, TP53 mutant cell-derived miR-769-3p and miR-21-3p synergistically activated fibroblasts in the lung; in turn, activated fibroblasts secreted TGF-β, further promoting the pulmonary metastasis of p53 R273H mutant cells, thus providing a basis for malignant development and metastasis.
Wild-type p53 can modulate the expression of several miRNAs via direct transcriptional regulation and indirect interaction with Drosha or Dicer, which are critical during miRNA biogenesis (35,36). Although most p53 hotspot mutations abolish the DNA-binding ability of p53,mutation of p53 leads to global changes in microRNA expression by binding to other mutant p53-interacting proteins, inhibiting the interaction among Drosha, p68 and primary miRNAs as well as reducing the level of Dicer (37).Mutant p53 upregulated the levels of miR-21-3p and miR-769-3p in our cell model and clinical samples. To date,there have been no relevant reports on the relationship between p53 and miR-21/miR-769, and the underlying mechanisms require further exploration.

Figure 5 TP53(R273H) mutation induces epithelial-mesenchymal transition (EMT) of HCT116 cells. (A) Heat map of overlapping genes between HCT116-TP53(WT) and HCT116-TP53(R273H) (MT) cells. Red/green indicates an increase/decrease in gene expression;duplicate biological repeats; (B) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of differentially expressed genes; (C) mRNA levels of ZEB1 and cell adhesion molecule-associated genes were determined using quantitative real-time polymerase chain reaction (qRT-PCR). Data represent theof relative mRNA levels; (D) Western blot analysis of EMT-associated genes in HCT116 cells with different TP53 statuses. β-actin was used as a loading control; (E) mRNA levels of CDH1, RhoA and SNAI2 were detected by qRT-PCR. *, P<0.05; **, P<0.01; ***, P<0.001.
Carcinoma-associated fibroblasts (CAFs) have been shown to exert protumorigenic effects, as they stimulate tumor proliferation, angiogenesis, invasion, and metastasis(38). These cells are considered myofibroblasts with myofibroblast-like gene and protein expression patterns and serve as the basic sources of growth factors,chemokines, extracellular matrix molecules, and matrixremodeling proteins in the tumor microenvironment. In addition to their paracrine signaling effects, CAFs could cooperate with other components of the tumor microenvironment, such as exosomes, to form a premetastatic niche. Exosomes could educate stromal cells in distant organs and even guide the organ tropism of tumor metastasis (6,8).
Exosomes contain large amounts of microRNAs. Herein,the p53 mutant increased the levels of miR-21-3p in tumor tissues and the subsequent output, which could have induced fibroblast activation. Therefore, combined with those in the literature, our results suggest that mature miR-21 in exosomes is a main factor in the activation of fibroblasts and promotion of pulmonary metastasis of colorectal cancer cells.
Furthermore, Smad7 has been confirmed as a target of miR-21-5p in breast phyllodes tumors and colorectal cancer (39,40). Although the seed sequence of miR-21-5p is different from that of our identified miR-21-3p, Smad7 is also predicted as a potential target of miR-21-3p according to the TargetScan and microRNA.org databases (41).Smad7 is an inhibitory Smad molecule that blocks activation of the TGF-β pathway. Our Western blot results demonstrated that Smad7 expression was inhibited by miR-21-3p, and a reverse correlation of Smad7 with miR-21-3p expression was observed in clinical samples.
In addition, this study showed that p53 mutation increased miR-769-3p levels in exosomes. Currently, very few studies on miR-769-3p and cancer have been reported.This microRNA is differentially expressed in pediatric gliomas and downregulated in plasma from epidermal growth factor receptor-tyrosine kinase inhibitor (EGFRTKI)-sensitive patients (42,43). In addition, miR-769-3p is an oxygen-dependent miRNA that directly inhibits NDRG1, resulting in the reduced proliferation and enhanced apoptosis of MCF-7 cells (44). Skp1 was predicted to be a potential target of miR-769-3p, and we found that miR-769-3p in exosomes could target Skp1 to significantly activate fibroblasts. Skp1 is an E3 ubiquitin ligase that is an indispensable component of and prerequisite for assembly of the multiprotein E3 ubiquitin ligase Skp1-Cullin1-F-box (SCF) complex (45). One known substrate of Skp1 is Smad3, a TGF-β receptor-regulated Smad (46); thus, Skp1 could be considered a negative regulator of TGF-β signaling. Therefore, miR-21-3p and miR-769-3p, which showed positively correlated expression, could synergistically enhance the TGF-β signal and promote the activation of fibroblasts to become myofibroblasts. In fact, we observed a synergistic effect of these miRNAs.
Activated myofibroblasts produce and secrete large amounts of TGF-β, and TGF-β further stimulates myofibroblasts to form a positive feedback loop. TGF-β also induces EMT of primary tumor cells, which further enhances their invasion and metastasis abilities. This cascading effect caused by p53 R273H mutant illustrates the communications between tumor and stromal cells, with exosomes acting as bridges between them (Figure 6E). We observed an EMT phenotype in HCT116-TP53(R273H)cells and xenograft tumor tissues and detected activation of fibroblasts in pulmonary metastasis.
Conclusions
Taken together, our data first indicate that p53 R273H mutation increases the secretion of miR-21-3p and miR-769-3p in exosomes to activate fibroblasts in the tumor microenvironment and lung tissues. This process facilitates formation of premetastatic niches to promote pulmonary metastasis of colorectal cancer cells. Furthermore, activated fibroblasts and mutant p53 promote acquisition of EMT phenotype by tumor cells to enhance their invasion and metastasis abilities. The identification of mutant p53-exosomal miR-21-3p/miR-769-3p-fibroblast-cytokine circuit deepens our understanding of the roles of mutant p53 in tumor metastasis in vivo, and miR-21-3p and miR-769-3p are potential predictive markers of pulmonary metastasis and candidate targets for therapeutic interventions.
Acknowledgements
This study was supported by grants from the National Key R & D Program of China (No. 2017YFC0906601, No.2017ZX10203205-003-001 and No. 2016YFC0901403),the National Natural Science Foundation (No. 81572840,No. 81572365, No. 81728015 and No. 81872033), the Nonprofit Central Research Institute Fund of CAMS (No.2018RC310011) and the CAMS Innovation Fund for Medical Sciences (No. 2016-I2M-1-001, No. 2017-I2M-3-005 and No. 2019-I2M-1-003) in China.
Footnote
Conflicts of Interest: The authors have no conflicts of interest to declare.
杂志排行

Chinese Journal of Cancer Research的其它文章
- Hexokinase II promotes the Warburg effect by phosphorylating alpha subunit of pyruvate dehydrogenase
- Adjuvant hormone therapy after radical prostatectomy in highrisk localized and locally advanced prostate cancer: First multicenter, observational study in China
- Cancer IgG, a potential prognostic marker, promotes colorectal cancer progression
- Burden of colorectal cancer in China, 1990-2017: Findings from the Global Burden of Disease Study 2017
- Impact of crizotinib on long-term survival of ALK-positive advanced non-small-cell lung cancer: A Chinese multicenter cohort study
- Correlation of radiotherapy with prognosis of elderly patients with hormone receptor-positive breast cancer according to immunohistochemical subtyping