亨廷顿舞蹈病一家系两例报道并文献复习
2019-07-12赵红刘赞华李淑敏蔺建文赵红玲王苏平
赵红,刘赞华,李淑敏,蔺建文,赵红玲,王苏平
亨廷顿舞蹈症(Huntington disease,HD)是一种以神经系统退行性改变为主要特征的常染色体显性遗传病,是由4号染色体IT15基因中CAG重复序列异常扩增所致,主要引起纹状体神经元退行萎缩,疾病症状缓慢进行性加重,典型症状包括舞蹈样不自主动作(晚期则运动能力逐渐丧失),精神障碍和进行性认知障碍。HD多发生于中年人,发病年龄40~50岁,但也偶见于儿童和青少年,称为青少年型亨廷顿症。世界范围内的HD发病率约为万分之一,各种人群均可患病,生存期为10~20年[1]。该病临床少见,易漏诊和误诊。本文对1例临床疑似HD的家系进行诊断分析,并结合文献,阐述HD的最新治疗进展,以加强对HD的认知。
1 病例简介
先证者,女,57岁,主因“面部及四肢不自主抽动3年”于2016-12-02就诊于大连市中心医院。患者3年前无诱因出现面部、眼肌、四肢及下颌部肌肉间歇性抽动,幅度小,持续数秒,反复发作,不影响日常生活,患者未察觉被家人发现后,于2013-09-02就诊于本院门诊,行颅脑CT检查未见确切异常,未予治疗。后患者上述症状逐渐加重,不自主抽动发作频繁,1 min数次,近半年患者自觉记忆力减退,远期记忆好,近期记忆差,但不影响正常生活,于2016-12-02就诊于本院门诊,行脑电图检查示广泛轻度异常,为进一步治疗收入神经内一科。查体:神清语利,计算力与近记忆力减退,理解力和判断力正常;眼动充分,未见眼震,双侧瞳孔等大等圆,直径2.5 mm,对光反射灵敏,双侧鼻唇沟对称,伸舌居中;四肢肌力正常,四肢肌张力对称减低,四肢针刺觉对称存在,面部及四肢间断细小抽动,双侧指鼻试验稳准,深感觉对称正常,双侧Babinski征阴性。辅助检查:肝功能检查、肾功能检查、血糖、血脂检查、离子、心肌酶、血尿常规均正常;甲状腺功能检查正常;肿瘤标志物和自身免疫检查(抗dsDNA、抗Sm抗体、抗ANA抗体)均正常。心电图示窦性心律,心电轴左偏,T波改变。颅脑磁共振成像(MRI)示脑内有小缺血性脱髓鞘改变,双侧基底核及皮质萎缩(见图1);双侧颞叶海马MRI示皮质萎缩。肝胆脾超声未见明显异常;颈动脉超声未见明显异常。经颅多普勒(TCD)示颅内外动脉血流未见明显异常。角膜色素(K-F)环阴性,外周血涂片未见棘红细胞;电生理示运动神经传导速度(MNCV)右侧胫神经腘窝部波幅降低,感觉神经传导速度(SNCV)正常,双侧尺神经F波正常,双侧胫神经H反射正常。简易智能精神检查量表(MMSE)评分27分,蒙特利尔认知评估量表(MoCA)评分19分(主要表现为执行功能和延迟回忆差)。
家系调查显示,该家系3代人共12例成员,其中3例发病,1例已故为先证者父亲(Ⅰ1)。Ⅰ160岁左右发病,肢体舞蹈样不自主扭动明显,疾病后期不能下床行走,后死亡。先证者哥哥(Ⅱ3)55岁左右发病,肢体舞蹈样不自主扭动,肢体运动不协调,幅度较大,目前使用针灸治疗。先证者儿子(Ⅲ1)和先证者哥哥的女儿(Ⅲ2),分别为28岁和30岁,无记忆力下降和肢体扭动。家系系谱图见图2。经患者及家属同意,采集先证者、先证者哥哥和先证者儿子的外周抗凝血5 ml,进行IT15基因检测,检测结果显示,先证者IT15基因CAG重复拷贝数为19/42,确诊为HD;先证者哥哥IT15基因CAG重复拷贝数为19/54,确诊为HD;先证者儿子IT15基因CAG拷贝数为19/22,未达到基因诊断标准。

图1 患者颅脑MRI显示脑部尾状核萎缩和侧脑室扩大Figure1 The cranial MRI showed atrophy of the caudate nucleus and enlargement of the lateral ventricle
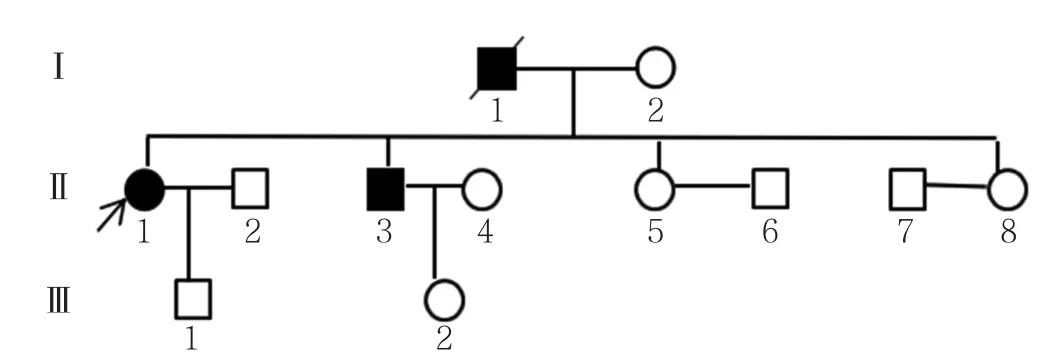
图2 HD患者的家系系谱图Figure2 Pedigree chart of HD family
本研究意义:
亨廷顿舞蹈症(HD)是一类严重影响人类健康而缺少任何根本性治疗手段的单基因遗传性神经退行性疾病。自HD致病基因IT15发现以来,近年来科学家们对HD的研究取得了重要突破,揭示了HD产生的遗传学机制、生化机制、分子机制及脑区特异性机制,将来有望针对这些机制研究出疗效更好的药物,为HD的根本性治疗奠定基础,并对包括HD在内的其他神经变性疾病的治疗产生积极的影响。本文对1例临床疑似HD的家系进行诊断分析,并结合文献,阐述HD的最新治疗进展,以加强对HD的认知。
2 讨论
1872年乔治·亨廷顿首次对HD进行了描述,指出其具有遗传性,并以其名字命名该病[2]。1983年GUSELLA等[3]通过遗传连锁分析将HD基因定位于4p16.3区域。1993年,HD协作研究组通过外显子扩增和cDNA克隆技术,确认CAG重复突变可导致HD发生,并发现了其致病基因IT15[4]。本文对1个家系的发病情况进行了调查,并对家系成员的IT15基因CAG重复拷贝数进行检测。
2.1 临床特点 HD是一种常染色体显性遗传的神经退行性疾病,由位于4号染色体4p16.3区域的IT15基因CAG三核苷酸重复序列异常扩增所致[4-5]。正常人群中CAG重复序列的长度为6~35个重复,如果CAG重复拷贝数超过40个,则会导致发病,出现异常运动症状;如果CAG重复拷贝数为36~39个,部分患者会发病,部分患者会继续保持无症状状态;CAG重复拷贝数为27~35个时属于可引起突变的等位基因,个体不会发病;但在减速分裂时不稳定,易发生扩展突变,使其后代患病,尤其是父系遗传时;CAG重复拷贝数<27个时为正常等位基因,个体正常,不引起疾病。CAG重复拷贝数>36个即为异常,重复拷贝数越高,发病年龄越早,症状越重[6]。HD平均发病年龄为40岁,青少年(<20岁)和老年(>70岁)也有发病。病因主要是家族遗传或者基因受到外部刺激而发生突变,只要双亲任一方遗传缺陷的基因,皆会表现出病征。HD的病理过程涉及多个方面,包括神经炎性反应、兴奋性氨基酸毒性作用、线粒体功能异常、转录失调等。病理机制仍未明确,认为CAG过度扩增,机体错误的制造一种名为“亨廷顿蛋白”的有害物质,这些异常蛋白质聚集成块,损害部分神经元,导致患者神经系统退化,并能发展为痴呆[7]。
HD主要累及纹状体和大脑皮质,具有高度的区域选择性。基底核运动通路受损引发运动过度—舞蹈样动作;大脑皮质受损导致患者认知功能障碍。神经影像学检查作为HD诊断的重要补充参考依据之一,颅脑CT及MRI示基底核及皮质萎缩,萎缩程度可能受CAG重复拷贝数的影响。本研究先证者颅脑MRI显示脑室、脑池稍大,脑沟稍宽,基底核区萎缩,以尾状核头部萎缩最明显,双侧侧脑室额角呈球形向外膨出,侧脑室尾状核区形成特征性的“蝴蝶征”,符合HD特征性影像学表现。本研究的先证者症状不典型,其肢体不自主抽动幅度小,不易察觉,没有明显的舞蹈样动作,虽有认知功能减退,未影响日常生活,易漏诊,经基因确诊为HD,其症状不典型考虑与其CAG重复拷贝数少有关。先证者哥哥症状典型,肢体舞蹈样扭动幅度大,认知功能减退明显,考虑与CAG重复拷贝数多有关。先证者儿子,30岁,无记忆力下降,无精神症状,无肢体扭动,基因检测未达到诊断标准。但有研究报道,1%的HD患者IT15基因检测显示阴性[8],故针对先证者儿子应长期随访,观察其症状发展。CAG重复序列与临床表现严重程度、影像学改变是否存在正相关,仍需进一步探讨。另外,对CAG重复拷贝数的检测除了对患者确诊外,更重要的是对患者及其子女提供咨询服务,以减少有害突变基因在家族中的遗传。
2.2 治疗与展望 目前仍未发现治疗HD的特异性药物,临床以对症治疗为主,针对病因、病机的治疗目前处于临床前阶段。丁苯那嗪是唯一被美国食品药品监督管理局(FDA)认可的用于治疗HD舞蹈动作的药物之一,可改善患者不自主运动症状,但可能会加重患者抑郁等精神症状[9]。抗帕金森药物可改善运动迟缓和肌强直,常见的有左旋多巴、金刚烷胺等。HD相关认知功能障碍尚无有效药物治疗[10],对于抑郁、焦虑及其他精神症状可给予抗抑郁(西酞普兰等)及抗精神病药物(奥氮平、利培酮等)改善症状。针对病因的治疗包括基因疗法和分子疗法,虽然针对病因的治疗难以实现,但针对HD发病机制的不同分子通路正在展开大量的研究,以期减少疾病发生。
线粒体是细胞内的多功能细胞器,不仅参与生物代谢能量产生过程,也能通过与内质网的相互作用调整钙稳态,产生自由基,参与细胞的死亡进程。线粒体在HD中发挥了重要作用,已有研究发现HD患者的细胞线粒体的形态和结构发生了明显的变化,这些异常变化可以导致神经元功能紊乱,参与HD的发生[11]。因此线粒体可以作为HD的治疗靶标。另有研究认为,HD是常染色体不稳定影响神经发生导致的发育性疾病[12],针对发育阶段进行基因调控成为新的治疗方法。
HD是由于γ-氨基丁酸(GABA)能神经元受损引起神经环路紊乱,进而导致患者出现运动功能障碍和认知能力丧失等一系列症状,GABA能神经元是分泌GABA的重要抑制性神经递质,对机体的多种功能有重要的调节作用[13]。如果脑内大量的GABA能神经元死亡,就会产生舞蹈样动作。将GABA能神经元移植到HD患者脑内,从而修复脑损伤环路,将成为HD治疗的新的方向。
神经营养因子为神经元提供营养性支持。研究发现,在3-硝基丙酸(3-nitropropionicacid,3-NP)致小鼠HD模型中,神经营养因子3(neurotrophin-3,NT-3)及其受体酪氨酸受体激酶C(tyrosine kinase receptors C,TrkC)mRNA及蛋白表达水平发生改变,NT-3通过TrkC受体调节纹状体区域的神经传递及突触可塑性[14]。神经营养因子对HD具有神经保护作用,通过遗传工程使细胞分泌神经营养因子并移植到纹状体[13],为治疗这一疾病带来了新的希望。
MicroRNA(miRNA)是长度为20~24个核苷酸的内源性小RNA,可以与靶标mRNA的3'UTR结合,调节蛋白质的表达转录。由于miRNA片段短,相对分子量小,能够通过血-脑脊液屏障,且在外周血中有较为稳定的表达,成为多种疾病诊断治疗及预后监测的新型生物标志物。近年来发现miRNA可以调节神经退行性相关基因的表达,参与多种神经退行性疾病如阿尔茨海默病、HD、帕金森病等发生[15]。有研究发现HD患者脑脊液中6种miRNA表达增高,且随着疾病风险增加而升高,这6种miRNA有望作为潜在的生物标志物[16]。以后可以研究如何特异地调节体内miRNA水平,通过降低miRNA的水平,从而推迟疾病的发作。
HD的治疗传统方法很难奏效,虽然基因治疗如外源性基因导入[17]、基因沉默[6,18-19]、细胞移植[20]等使 HD 的治疗有了新的方向,然而这些方法尚处于探索之中,任重而道远,期待更多研究为HD的治疗带来新的曙光。有关HD进展的生物标志物的研究正在不断进行,HD的治疗前景仍很乐观。
作者贡献:赵红、刘赞华进行文章的构思与设计,文献/资料整理;李淑敏、赵红玲进行文章的可行性分析;蔺建文、赵红玲进行文献/资料收集;赵红撰写论文;李淑敏、蔺建文进行论文的修订;赵红、王苏平负责文章的质量控制及审校,对文章整体负责,监督管理。
本文无利益冲突。
