卵泡抑素样蛋白4通过活化T细胞核因子抑制病理性心肌肥厚的发生发展
2019-07-01鲁悦新石红杰欧阳珊孔祥杰胡宇峰姬燕晓李红良
鲁悦新 石红杰 欧阳珊 孔祥杰 胡宇峰 姬燕晓 李红良
病理性心肌肥厚是心力衰竭和猝死发生的重要原因[1],引起心肌肥厚的原因很多,临床上主要受遗传因素、高血压、内分泌、生活习惯等复杂因素共同影响。其中高血压是临床常见的心肌肥厚诱因之一,高血压性心脏病常伴随一系列并发症,包含左室肥厚、左房扩大、冠状动脉疾病、充血性心力衰竭等[2]。临床上很多治疗高血压的药物同样对心肌肥厚有很好的治疗作用[3]。研究表明,许多调控高血压的基因也参与调控心肌肥厚发生[4]。卵泡抑素样蛋白4(follistatin-like 4,FSTL4)是TGF-β超家族抑制剂follistatin基因家族的一员,广泛表达于心脏、肺、肾、睾丸、神经元、平滑肌和肠上皮中[5-6]。此前的一项研究,通过对高血压患者及其家族的基因图谱进行单核苷酸多态性分析,发现FSTL4的突变可能是影响高血压的一个关键诱因[6]。此外,研究发现FSTL4是肌肉发育的关键调节剂,与卒中风险增加相关[6-7]。因此,笔者利用FSTL4基因敲除小鼠和FSTL4过表达的心肌细胞来深入探究FSTL4在心肌肥厚中的作用与机制。
1 材料与方法
1.1 动物实验材料与方法
1.1.1实验动物 野生型(WT组)小鼠8只,FSTL4基因敲除(KO组)小鼠8只,体重24~26g、10周龄雄性小鼠。KO小鼠购买自RIKEN BioResource Research Center(RBRC04952),饲养于武汉大学模式动物研究所SPF级实验动物屏障设施。
1.1.2病理性心肌肥厚模型的建立 WT和KO组小鼠均进行主动脉弓缩窄手术(TAC)[8-9],构建压力超负荷诱导的左室心肌肥厚模型。腹腔注射3%戊巴比妥钠麻醉小鼠,沿第2~3肋间水平切开皮肤,依次分离肌肉及软组织,暴露出胸乳突肌,然后用直尖头镊提起左侧锁骨,用解剖剪紧贴胸骨剪断小鼠右侧锁骨,游离主动脉弓(无名动脉和左颈总动脉分叉处),将7-0手术缝线穿过主动脉,将26G垫针及主动脉一起结扎,抽出针头造成主动脉70%缩窄,然后依次缝合切口。之后将小鼠以俯卧位放入恒温箱中至苏醒后,将小鼠放入装有高压灭菌过的垫料、饲料和饮用水的饲养笼内,于饲养室继续饲养观察。
1.1.3超声检测 术后4周用异氟烷麻醉小鼠后,对左胸前区剃毛,小鼠取左侧卧位或仰卧位,在剃毛区均匀涂抹超声耦合剂。采用高频超声诊断仪,频率设置为15 MHz,选取标准左室乳头肌短轴切面进行超声检测,测量并计算左室舒张末期内径(LVEDd)、左室收缩末期内径(LVESd)、短轴缩短率(FS)及射血分数(EF)。
1.1.4组织病理学检测 超声检测结束后,称重后处死小鼠,迅速取出心脏和肺脏,分别称重并记录。剪开小鼠后肢胫骨处皮肤,测量并记录胫骨长度。计算心重体重比(HW/BW)、肺重体重比(LW/BW)、心重与胫骨长度的比值(HW/TL),评价心脏重量的增大程度。称重之后将心脏放入10%的福尔马林中固定24 h,再进行脱水、石蜡包埋、切片,天狼猩红染色(PSR)和苏木精-伊红染色(H&E)。
1.1.5实时荧光定量聚合酶链式反应(qPCR) 用TriPure Isolation Reagent(11667165001,Roche)提取心脏左心室的RNA,用逆转录试剂盒(4897030001,Roche)反转录成cDNA,加入荧光染料SYBR Green(04887352001,Roche)标记扩增产物进行qPCR实验,检测心肌肥厚基因心钠肽(Anp)、脑钠肽(Bnp)和心脏β-肌球蛋白重链(Myh7),心肌纤维化基因I型胶原(Collagen Iα )、Ⅲ型胶原(Collagen Ⅲ)和结缔组织生长因子(Ctgf)的表达。所用引物由擎科生物科技有限公司进行化学合成,序列信息如下表:

表1 qPCR引物序列信息
1.1.6蛋白质印迹(Western blot)检测 用眼科剪迅速将心脏左室样本剪碎后放入EP管中,在干冰上操作,称重并记录样本重量。向每个EP 管中加入4颗钢珠和RIPA裂解液(100 μl/10 mg),放入研磨仪中研磨组织,设置研磨参数:频率30 Hz,时间90 s。待研磨结束后取出钢珠,使用超声裂解仪裂解组织细胞,频率5K Hz,时间120 s。裂解完成后离心获得蛋白提取液,使用BCA 法(23225,Thermo Fisher Scientific)调平蛋白浓度,将蛋白样品变性后进行SDS-PAGE凝胶电泳。电泳后将蛋白从凝胶转膜至PVDF膜,对PVDF膜使用5%的脱脂牛奶室温封闭1 h,4℃孵育一抗过夜,室温孵育二抗1 h,于Bio-Rad ChemiDocTMXRS+化学发光成像系统进行显影。实验中使用的一抗包括:FSTL4(ab214925,ABCAM)、GAPDH(2118,CST)、Flag(M185-3L,MBL)。
1.2 细胞实验材料与方法
1.2.1H9C2细胞免疫荧光染色 H9C2细胞购买自美国模式培养物集存库(ATCC),利用慢病毒感染H9C2细胞构建FSTL4过表达的稳定细胞系。对细胞系饥饿12 h处理后,用1 μmol/L血管紧张素Ⅱ(AngⅡ)或对照液PBS刺激48 h,刺激后使用4%多聚甲醛固定细胞,将细胞置于0.1%Triton X-100中透化5 min,室温下用8%羊血清封闭1 h,加α-actinin(05-384,Merck Millipore,1∶100)室温孵育2 h进行染色,PBS漂洗后滴加荧光二抗(IgG[H + L]二抗,A21202,Invitrogen,1∶200)室温孵育1 h,PBS漂洗后加DAPI封片,于荧光显微镜下进行拍照。使用Image-Pro Plus 6.0软件测量细胞的表面积。
1.2.2双荧光素酶报告基因检测 对H9C2细胞同时表达对照Flag或FSTL4质粒、NFAT-luc、对照Actin-luc,18 h后用2μmol/L AngⅡ或对照液PBS刺激细胞12 h,刺激完成后裂解细胞。吸取20 μl细胞裂解液和100 μl荧光素酶测试试剂Ⅱ(LARII)混匀,立即检测萤火虫荧光素的荧光值,加入100 μl STOP& Glo Reagent试剂混匀,立即检测海肾荧光素的荧光值,荧光素酶的表达量与转录因子FSTL4对活化T细胞核因子(NFAT)的转录活性作用强度成正比。
1.3 统计学方法
数据结果通过平均数±标准差来表示,采用SPSS 19.0软件进行统计学分析,两组之间均数比较使用独立样本t检验,多组间均数比较使用单因素方差分析,以P<0.05为差异有显著性。
2 结果
2.1 两组小鼠实验结果
2.1.1两组超声指标及心肌肥厚程度的比较 Western blot结果显示KO小鼠心脏组织中FSTL4基因完全敲除(图1A)。TAC手术4周后,KO组小鼠的LVEDd和LVESd均显著高于WT组;而EF和FS显著低于WT组(表2)。TAC手术4周后KO组小鼠的HW/BW、LW/BW和HW/TL均显著高于WT组(表3)。

A:两组小鼠心脏组织中FSTL4表达水平;B:TAC手术4周后两组小鼠心肌肥厚和心肌细胞肥大程度
图1两组小鼠FSTL4蛋白表达和心肌肥厚程度的比较

表2 两组超声指标的比较
注:与WT组比较,*P<0.05

表3 两组HW/BW、LW/BW、HW/TL的比较
注:与WT组比较,*P<0.05
HE染色结果显示KO组小鼠心脏体积明显增大,心肌细胞面积显著增加(图1B,表4)。TAC手术4周后,qPCR结果显示KO组小鼠的Anp、Bnp和Myh7水平都高于WT组(表5)。

表4 两组心肌细胞面积的比较
注:与WT组比较,*P<0.05
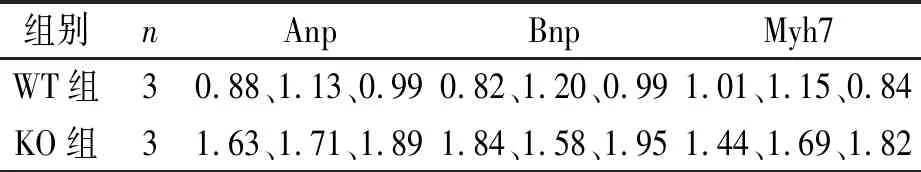
表5 两组心肌肥厚基因表达量的比较
注:因n=3,未进行统计学处理,但KO组3个数据均高于WT组
2.1.2两组心肌纤维化程度的比较 PSR染色结果显示,KO组小鼠心脏组织血管周纤维化和间质纤维化程度加重(图2),统计学分析表明KO组小鼠心肌的胶原纤维含量显著升高(表6)。qPCR结果显示,KO组小鼠的心脏纤维化指标Collagen Iα、CollagenⅢ和Ctgf都升高,显示出严重的纤维化(表7)。
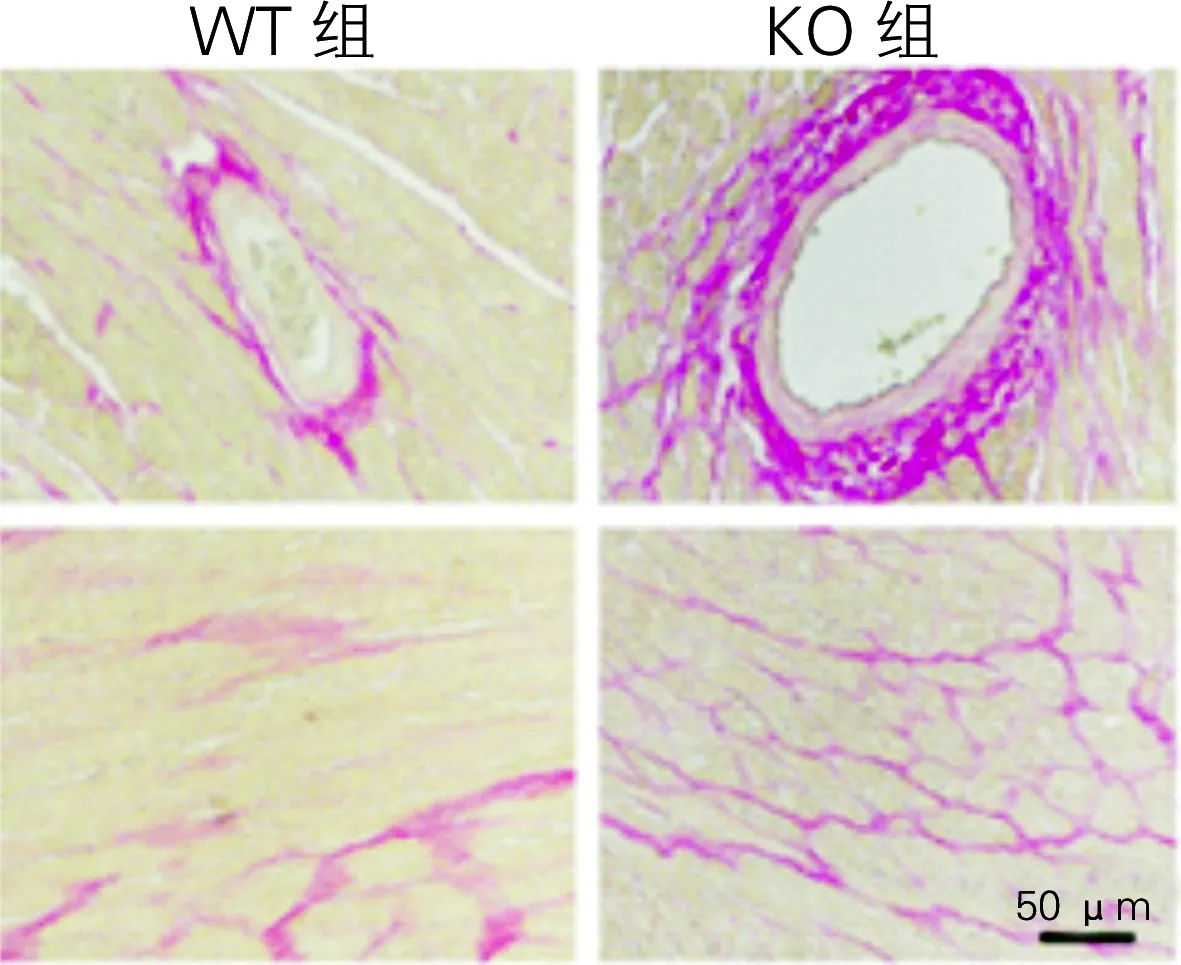
TAC手术4周后两组小鼠心肌纤维化程度

组别n胶原百分比(%)WT组52.03±0.78KO组55.19 ±2.50∗
注:与WT组比较,*P<0.05

表7 两组心肌纤维化基因表达量的比较
注:因n=3,未进行统计学处理,但KO组3个数据均高于WT组
2.2 H9C2细胞实验结果
2.2.1FSTL4对心肌细胞肥大的影响 Western blot结果显示FSTL4稳定细胞系中FSTL4的蛋白表达水平增加情况(图3A)。免疫荧光染色结果显示AngⅡ刺激导致心肌细胞肥大,FSTL4过表达能够抑制AngⅡ刺激导致的心肌细胞肥大(图3B、表8)。qPCR结果显示过表达FSTL4能够显著降低心肌肥厚指标Anp和Myh7的表达(表9)。
2.2.2FSTL4对NFAT转录活性的影响 双荧光素酶报告基因检测结果显示,AngⅡ刺激H9C2细胞后活化T细胞核因子(NFAT)的转录活性显著上升,过表达FSTL4能够显著抑制AngⅡ刺激诱导的NFAT转录激活(表10)。
3 讨论
本研究采用FSTL4-KO小鼠为研究对象,在主动脉弓缩窄手术4周后检测心脏重量、通过病理分析

A:H9C2细胞中FSTL4的过表达效率;B:AngII刺激后对照和过表达FSTL4的H9C2心肌细胞肥大程度

图3 FSTL4过表达对心肌细胞肥大的影响
注:与Flag PBS组比较,*P<0.05;与Flag AngII组比较,#P<0.05
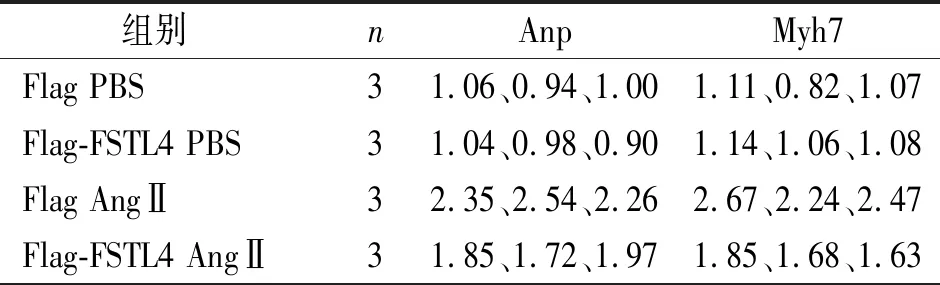
表9 不同组别心肌肥厚基因表达量的比较
注:因n=3,未进行统计学处理,但Flag AngⅡ组3个数据均高于Flag PBS组,Flag-FSTL4 AngⅡ组数据均低于Flag AngⅡ组

表10 不同组别NFAT转录活性的比较
注:因n=4,未进行统计学处理,但Flag AngⅡ组3个数据均高于Flag PBS组,Flag-FSTL4 AngⅡ组数据均低于Flag AngⅡ组
和超声检测,结合实时荧光定量PCR检测心肌肥厚和心脏纤维化标志基因变化,全面考察了FSTL4对压力超负荷诱导的病理性心肌肥厚的功能和作用。结果显示FSTL4敲除之后,心肌细胞增大,心肌肥厚程度加重,心功能恶化,心肌纤维化程度加重,表明FSTL4敲除进一步促进了病理性心肌肥厚和心脏重构的发生发展。此外,采用FSTL4过表达的H9C2细胞为研究对象,并使用AngⅡ刺激诱导心肌细胞肥大模型。结果显示过表达FSTL4显著降低AngⅡ诱导的心肌细胞体积增大和心肌肥厚相关指标的表达,提示过表达FSTL4可抑制心肌肥厚的发生。
据报道,心肌肥厚的发生受多种复杂细胞信号通路调控,包含丝裂原活化蛋白激酶、磷脂酰肌醇-3-激酶/蛋白激酶 B、蛋白激酶 C和钙调神经磷酸酶(Calcineurin)/激活NFAT等信号通路[10-12]。这些复杂的信号网络,通过感知上游刺激,调控基因转录、线粒体功能、内质网应激、活性氧释放、炎症反应等一系列分子事件,调控心肌细胞中大量蛋白质合成和表型转化[13]。FSTL4 预测为一个分泌蛋白,结构分析表明其由一个类卵泡抑素结构域、一个钙离子结合结构域和两个类免疫球蛋白结构域组成。FSTL4可影响小鼠视网膜发育[14],此外,FSTL4通过调控脑源性神经营养因子的成熟和外泌来调控小鼠轴突分支[15]。
本研究显示FSTL4抑制NFAT转录活性,提示FSTL4很可能作为一个钙离子结合蛋白,通过调控Ca2+-Calcineurin-NFAT信号通路,影响下游NFAT信号通路的激活。Ca2+增加可以激活钙调神经磷酸酶Calcineurin,与转录因子NFAT结合,通过去除NFAT N端保守的丝氨酸残基对其进行去磷酸化,导致NFAT向细胞核易位,激活心肌肥厚前体基因的表达。据报道,转录因子NFAT在激活后,可直接调控心肌肥厚标志物BNP的表达[16]。本研究中小鼠敲除FSTL4后,BNP表达显著性上升,提示FSTL4可能通过Calcineurin-NFAT信号调控心肌肥厚和心脏重构发生。
