固相萃取-超高效液相色谱-串联质谱法同时测定保健食品中21种非法添加化合物
2019-06-24徐敦明赖国银伊雄海邓晓军厦门海关技术中心福建厦门606上海海关动植物与食品检验检疫技术中心上海005中国检验检疫科学研究院食品安全研究所北京00
徐敦明, 赖国银*, 陈 燕, 罗 超, 伊雄海, 邓晓军, 冯 峰, 张 峰(.厦门海关技术中心,福建 厦门 606;.上海海关动植物与食品检验检疫技术中心,上海 005;.中国检验检疫科学研究院食品安全研究所,北京 00)
近年来,随着人民生活水平的提高和保健食品加工产业的发展,人们对保健食品的需求量越来越大。由国家食药监总局及地方食药监局2016~2017年食品抽检公告可知,尽管保健食品总体合格率很高,达98.3%,但仍有40余例保健食品非法添加案例,主要集中在减肥、辅助降血糖、免疫调节类产品,其中绝大多数为假冒产品。一些不法厂商为达到产品宣称的功效,在保健食品中直接添加化学药物,消费者若长期食用该类保健食品,身体将产生不良反应。
原国家食品药品监督管理总局已发布了对减肥类、降脂类、降压类、降糖类保健食品中违法添加化学药物的补充检测方法,编号分别为2006004、2009029、2011008、2012005、201300及食药监办许(2010)114号,大部分的检测方法采用薄层色谱法、高效液相色谱法和液相色谱-串联质谱法,样品前处理为超声提取后直接上机测定。然而保健食品中成分较多,基质复杂,基质效应(ME)强且对仪器的抗污染能力要求较高。目前用于减肥类、降脂类、降糖类、降压类保健食品中非法添加药物的检测方法主要有液相色谱法[1-4]、液相色谱-串联质谱法[5-21]、超高效液相色谱-四极杆-飞行时间质谱法[22]、拉曼光谱法[23]、薄层色谱法[24]等。其中,液相色谱-串联质谱法因具有高选择性、高灵敏度的优点在保健食品中非法添加药物的定性及定量分析中得到了广泛的应用。保健品有口服液、片剂、硬胶囊和软胶囊等不同剂型,特别是胶囊剂型,由于制剂辅料的加入,使样品基质更加复杂,影响了方法的灵敏度和抗干扰性[21]。因此,选择合适的前处理净化方法显得尤为重要。
针对4种不同剂型,本文重点对提取溶剂、净化柱及洗脱溶剂进行了研究论证。为打击不同功效保健食品中交叉添加化学药物的行为,减少样品在测定过程中对仪器带来的污染,降低基质干扰对测定的干扰,本文通过优化净化过程,建立了超高效液相色谱-串联质谱法,可以同时筛查测定减肥类、降脂类、降糖类、降压类保健食品中21种非法添加药物。
1 实验部分
1.1 仪器、试剂与材料
TQD超高效液相色谱-串联质谱仪,美国Waters公司;P 300H超声波清洗器(超声功率380 W),德国Elma公司;分析天平,瑞士梅特勒公司;涡旋混合器,德国IKA公司;氮吹仪,美国Organomation公司。
甲醇、乙腈,色谱纯,德国Merck公司;乙酸铵,色谱纯,美国Sigma公司。
标准品:麻黄碱(ephedrine)、芬氟拉明(fenfluramine)、N,N-双去甲基西布曲明(N,N-didesmiehylsibutramine)、N-单去甲基西布曲明(N-monodesmiehylsibutramine)、西布曲明(sibutramine)、苯乙双胍(phenformin)、甲苯磺丁脲(tolbutamide)、格列齐特(gliclazide)、吡咯列酮(pioglitazone)、格列波脲(glibornuride)、格列吡嗪(glipizide)、瑞格列奈(repaglinide)、格列美脲(glimepiride)、格列本脲(glibenclamide)、格列喹酮(gliquidone)、可乐定(clonidine)、利血平(reserpine)、去羟基洛伐他汀(dehydro lovastatin)、美伐他汀(mevastatin)、洛伐他汀(lovastatin)、辛伐他汀(simvastatin)均购于中国食品药品检定研究院。混合型阴离子(PAX)固相萃取柱(60 mg/3 mL),美国Agilent公司;亲水亲脂型(HLB)固相萃取柱(60 mg/3 mL),美国Waters公司。
1.2 样品前处理
颗粒、片剂样品:取适量,研细;胶囊样品:取内容物;液体样品:取1 g,加入1 mL饱和氯化钠溶液。
称取0.5 g(精确至0.000 1 g)处理过的供试样品,置于50 mL离心管中,加入10 mL乙腈,涡旋混匀后超声提取15 min,以5 000 r/min离心10 min,移取乙腈层至另一离心管中;残渣再用10 mL乙腈超声提取15 min,以5 000 r/min离心10 min;合并乙腈层,氮吹约至0.5 mL后,加入10 mL水涡旋,待净化。
将HLB固相萃取柱接于固相萃取装置上,依次用5 mL甲醇、5 mL水活化小柱,用5 mL 5%(体积分数,下同)甲醇水平衡后,将待净化的样品溶液加入小柱内,控制流速1滴/s,待样品溶液流尽后,用4 mL 5%甲醇水溶液淋洗小柱,最后用5 mL 90%甲醇水溶液洗脱小柱,收集洗脱液至5 mL容量瓶中,定容至刻度,经0.22 μm混合型滤膜过滤后作为测定液。
1.3 分析条件
色谱柱为Waters BEH C18柱 (100 mm×2.1 mm,1.7 μm);柱温为30 ℃;流动相:A为10 mmol/L乙酸铵水溶液,B为甲醇;流速为0.3 mL/min。梯度洗脱程序:0~2.0 min,70%A;2.0~4.0 min,70%A~40%A;4.0~8.0 min,40%A~30%A;8.0~10.5 min,30%A~70%A;10.5~14.0 min,70%A。进样量为2 μL。
离子源为电喷雾电离(ESI)源;扫描方式为正离子扫描;检测模式为多反应监测(MRM)模式;去溶剂气流量为900 L/H;去溶剂气温度为650 ℃。
21种目标化合物的保留时间、母离子、子离子、锥孔电压(CV)、碰撞电压(CE)等其他质谱参数见表1。

表1 21种目标化合物的质谱参数Table 1 MS parameters of the 21 target compounds

表1 (续)Table 1 (Continued)
* Quantitative ion.
2 结果与讨论
2.1 色谱条件的优化
实验分别考察了甲醇-水、乙腈-水作为流动相体系时的分离效果,发现用甲醇-水时,降脂类等化合物的响应明显增强,且各化合物之间分离效果较好。此外,在水中加入10 mmol/L乙酸铵后明显能改善化合物的峰形。因此最终选用10 mmol/L乙酸铵水溶液-甲醇作为流动相体系,此时21种目标化合物分析时间较短,分离效果好,响应高。
2.2 质谱条件的优化
采用直接进样方式对21种目标化合物进行一级质谱分析,得到各自的母离子。对目标化合物进行二级质谱扫描,得到物质的子离子信息,确定目标化合的定量、定性离子对。通过优化各物质的锥孔电压、碰撞能量,确定各目标化合物的质谱参数(见表1)。图1为代表性药物麻黄碱、芬氟拉明、吡咯列酮和美伐他汀(100 μg/L)的多反应监测色谱图。
2.3 提取方法的优化
2.3.1提取溶剂的优化
实验对比了甲醇、乙腈两种提取溶剂对提取效率的影响。结果发现,两种溶剂对目标化合物的提取效率无显著性差异,考虑到口服液样品中存在水,而甲醇与水混合后不能有效分层,故最终选择乙腈作为提取溶剂。
2.3.2超声时间的优化
本实验对比了不同超声时间(10、15、20、25和30 min)对提取效率的影响。结果显示,超声时间对提取效率无显著性差异,从节约、环保的角度考虑,结合目前国家食品药品监督管理总局颁布的对各化合物补充检验方法中的提取时间,本实验确定15 min为样品超声时间。

图1 麻黄碱、芬氟拉明、吡咯列酮和美伐他汀(100 μg/L)的多反应监测色谱图Fig.1 Chromatograms of ephedrine,fenfluramine, pioglitazone and mevastatin (100 μg/L)in multiple reaction monitoring (MRM)mode
2.3.3净化方法的优化
针对21种目标化合物结构式中存在的多种官能团,如季铵离子、苯环,实验考察了QuEChERS(900 mg Mg2SO4、150 mgN-丙基乙二胺(PSA)、15 mg石墨化炭黑(GCB))、PAX固相萃取柱、HLB固相萃取柱对样品的净化效果。结果显示,采用QuEChERS及PAX固相萃取柱时,某些目标化合物的回收率不够理想,而采用HLB固相萃取柱时,目标化合物的回收率均较好。故实验选用HLB固相萃取柱作为净化小柱。
以HLB固相萃取柱作为净化小柱,本实验考察了不同体积分数的甲醇水溶液作为洗脱液时的洗脱效果。将21种目标化合物的水溶液上样,然后分别用体积分数为0%、5%、10%、20%、30%、40%、50%、60%、70%、80%、90%、100%的甲醇水溶液洗脱,收集洗脱液上机测定,考察各目标化合物的洗脱效果(见图2)。实验表明,当甲醇体积分数在70%以下时,仅有麻黄碱、芬氟拉明、可乐定和利血平被洗脱下来;当以90%甲醇水溶液为洗脱溶剂时,目标化合物的整体回收率最好,大部分物质的回收率>80%。因此实验选择90%甲醇水溶液作为洗脱溶液。
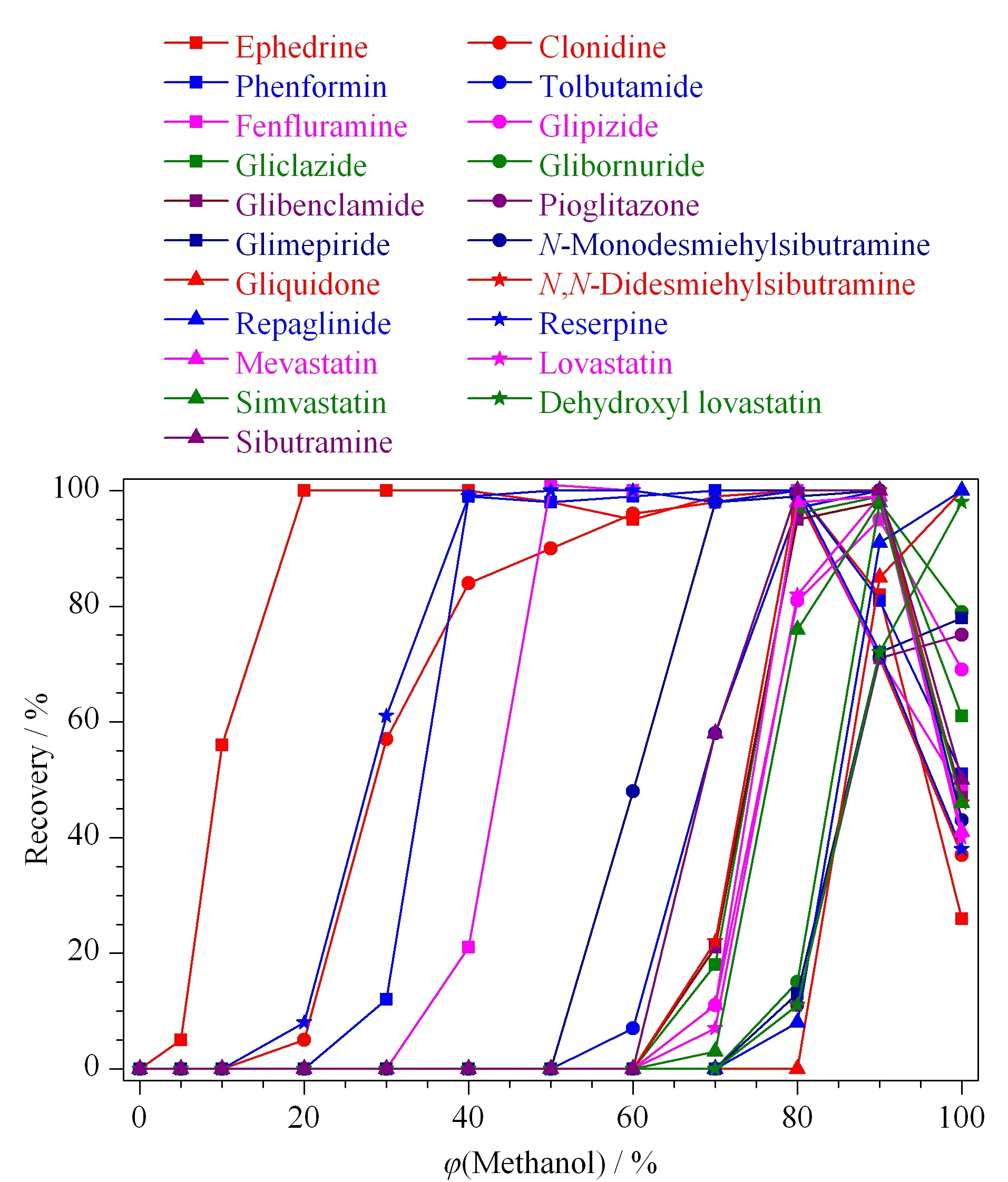
图2 甲醇的体积分数对21种化合物(100 μg/L)回收率的影响Fig.2 Effect of volume of percentage of methanol on the recoveries of the 21 compounds (100 μg/L)
2.4 方法学评价
2.4.1基质效应
与其他分析手段不同的是,质谱分析尤其是以电喷雾电离源为离子源的质谱分析可能会存在基质效应。保健食品多采用天然材料,基质中的杂质会对目标化合物的分析产生一定的干扰,影响分析的准确度、灵敏度和精密度。本实验选取了口服液、片剂、硬胶囊和软胶囊4种具有代表性的空白基质溶液,与定容溶剂分别配制相同浓度的标准溶液。采用公式ME=(基质匹配曲线的斜率/标准曲线的斜率-1)×100%计算基质效应。基质效应为负值时表示存在基质抑制效应;基质效应为正值时表示存在基质增强效应。结果表明,大部分化合物在这几种基质中都有不同程度的基质抑制效应(见表S1,详见http://www.chrom-China.com,下同)。特别是吡格列酮和去羟基洛伐他汀的基质抑制效应较大,均大于20%。为了消除基质效应带来的影响,本文采用基质匹配曲线进行线性观察和含量测定。
2.4.2方法的线性范围、检出限和定量限
为考察样品基质的影响,实验分别使用甲醇和口服液、片剂、硬胶囊、软胶囊空白基质溶液配制系列标准溶液,以色谱峰的峰面积对待测成分的质量浓度绘制标准曲线。结果表明,在考察的范围内,各种基质中21种非法添加化合物均呈良好的线性关系,相关系数均≥0.995(见表S1)。
通过向4种剂型的阴性样品中添加21种待测成分,考察方法的检出限(S/N≥3)和定量限(S/N≥10),不同基质的检出限为3~160 μg/kg,定量限为10~500 μg/kg(见表2)。
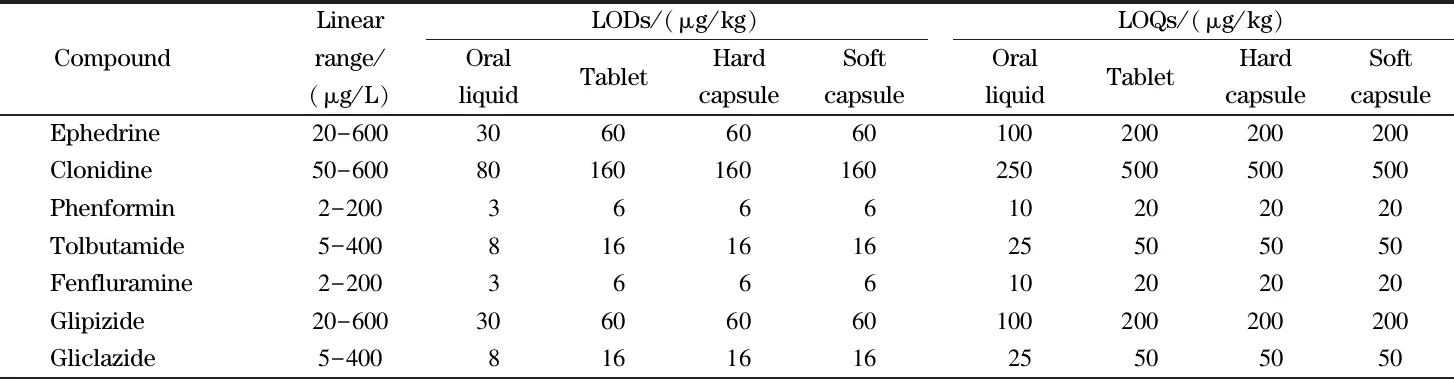
表2 21种目标化合物的线性范围、检出限和定量限Table 2 Linear ranges,limits of detection (LODs)and limits of quantification (LOQs)of the 21 target compounds

表2 (续)Table 2 (Continued)
2.4.3回收率和精密度
按照1.2节的前处理方法,向空白基质中分别加入低、中、高(100、500和1 000 μg/kg)3个水平的加标量,进行加标回收率和精密度试验,结果见表3。4种剂型中21种化合物的平均回收率为61.8%~109.3%,相对标准偏差为1.6%~14.7%。方法的加标回收率和精密度都均符合残留检测的要求。
2.5 实际样品检测
采用本文方法对市售常见的保健食品进行检测,其中口服液、片剂、硬胶囊和软胶囊各5份,涵盖减肥类、降脂类、降糖类和降压类。结果表明,在一个口服液产品中检出苯乙双胍(1.2 mg/kg);在一个片剂中检出西布曲明(10.6 mg/kg)和格列美脲(504.3 mg/kg)。
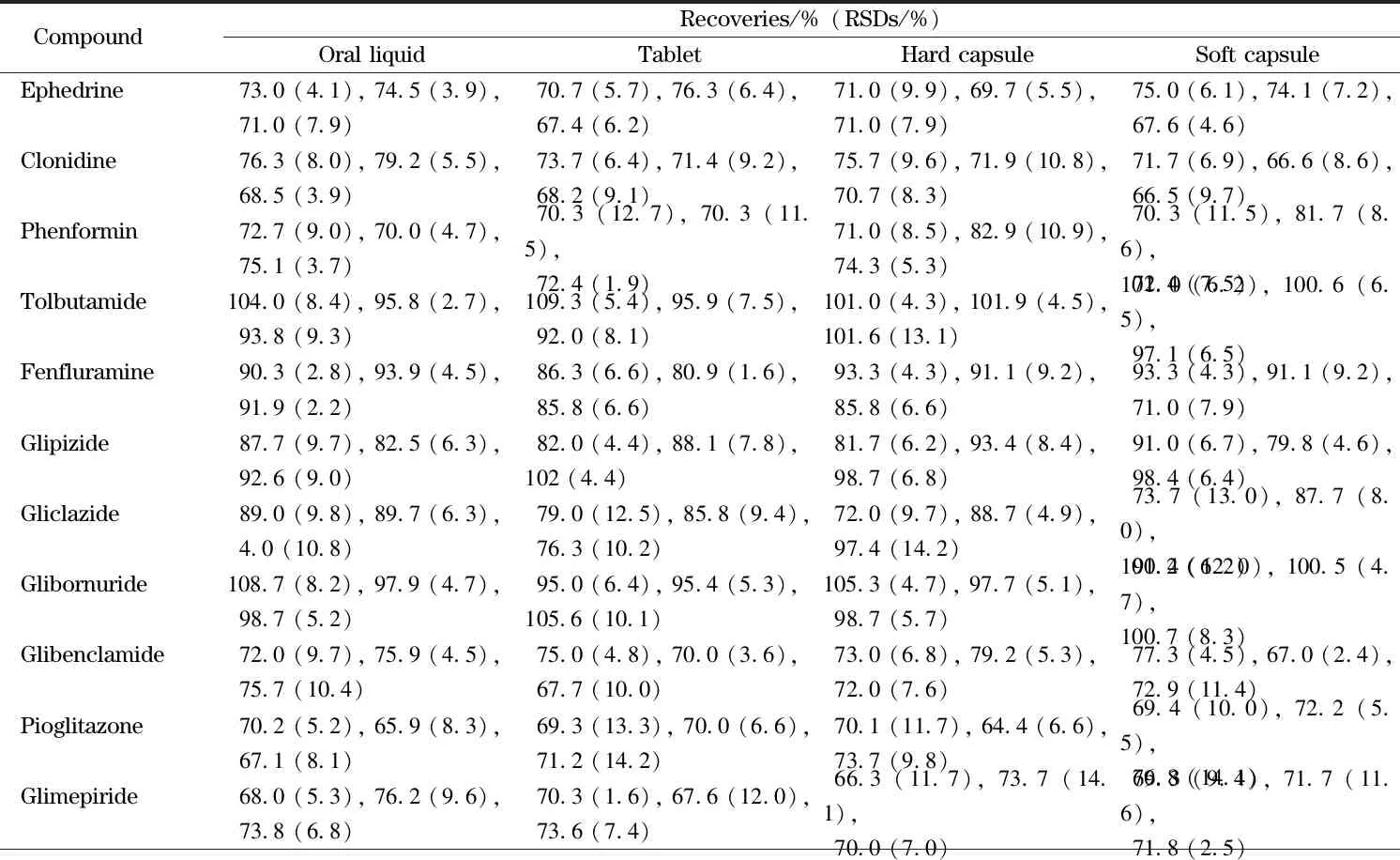
表3 4种剂型中21种目标化合物的加标回收率和相对标准偏差(n=6)Table 3 Spiked recoveries and relative standard deviations (RSDs)of the 21 target compounds in the four dosage forms (n=6)

表3 (续)Table 3 (Continued)
Recoveries and RSDs from left to right represented the results of three spiked levels of 100,500,and 1000 μg/kg.
3 结论
本文建立了超高效液相色谱-串联质谱同时测定减肥类、降脂类、降压类、降糖类保健食品中21种非法添加药物的方法。该方法能够满足同时检测多类保健食品中非法添加药物的要求,可以为保健食品非法添加的检测工作提供有力的技术支持。
