自噬与肿瘤EGFR抑制剂耐药的研究进展
2019-06-06杨超波李冠武
杨超波,童 欣,李冠武
(汕头大学医学院肿瘤分子生物学开放实验室,广东 汕头 515041)
表皮生长因子受体(epidermal growth factor receptor,EGFR)是一个带有酪氨酸激酶活性的跨膜蛋白.研究表明它在许多肿瘤中过表达,异常的EGFR 信号通路在肿瘤发生、进展和转移中发挥着重要作用.EGFR 的抑制剂被广泛用于治疗携带EGFR 突变的肺癌患者.目前应用于临床的EGFR抑制剂包括两种类型,单克隆抗体和酪氨酸激酶抑制剂,前者通过竞争性结合EGFR 的胞外域来抑制受体的二聚化或促进受体的内化;后者通过竞争性结合ATP 来抑制EGFR 酪氨酸激酶结构域的活性.然而,经过一段时期治疗后,绝大多数患者都会产生耐药.因此,克服耐药是临床治疗肿瘤的一大重要目标.自噬是细胞处于营养缺乏或应激状态时的一种自我保护机制.近年来越来越多的研究显示,自噬在肿瘤EGFR抑制剂耐药中发挥重要作用.EGFR抑制剂能诱导自噬,而自噬在肿瘤细胞中既可作为保护因素来介导耐药,又可作为毒性因素来延缓耐药.因此,调节自噬将为克服肿瘤患者EGFR抑制剂耐药提供一可行思路.本文对自噬与EGFR抑制剂耐药的最新研究进展进行综述.
1 自噬的机制与调节
大自噬(在这篇综述中指的是自噬)是一个进化上古老和高度保守的新陈代谢过程,它涉及双层膜囊泡的形成,该囊泡吞噬细胞内蛋白和细胞器并把它们运输到溶酶体中降解.当细胞遭受各种化学或物理因素作用例如营养缺乏、低氧、活性氧(ROS)积累和DNA破坏等,自噬被诱导以应对外界压力.自噬的过程可被划分为这5 个阶段:自噬启动、吞噬泡形成、吞噬泡成熟、自噬体融合和货物分解,其中,前三个阶段需要各种自噬相关基因(ATGs)编码的蛋白参与.自噬的启动开始于ULK1(也称为ATG1)复合物(包含有ULK1、ULK2、ATG13、FIP200 和ATG101),它活化III 型PI3K 复合物,该复合物包含有VPS34(也称为PIK3C3)、ATG14、UVRAG 和AMBRA1,所有这些均被公认的肿瘤抑制因子Beclin1 所结合,此过程即形成吞噬泡.随后,吞噬泡内发生各种蛋白的组装.ATG7 激活ATG12,后者通过ATG10 共价结合于ATG5.ATG5 与ATG16 结合,最终形成出现在吞噬泡膜外表面上的Atg12-Atg5-Atg16 复合物,该复合物有助于吞噬泡的伸长与闭合.ATG4剪切微管相关蛋白轻链3(LC3)形成LC3-I,LC3-I 暴露的剪切位点被ATG7 活化并转移到ATG3.LC3-I-ATG3 复合物共价结合磷脂酰乙醇胺(PE)形成LC3-II,后者整合入自噬泡膜上形成自噬体.最后,自噬体与溶酶体接触并融合,成为自噬溶酶体,其中的内容物被降解、大分子前体物质被循环利用或用于促进新陈代谢[1].自噬诱导过程与细胞内多条控制细胞增殖或凋亡的分子通路有交互作用.在这些通路中,PI3K/AKT/mTOR 是调节自噬最重要的信号通路,它也是人类肿瘤最常见和最典型的生存机制.此外,作为细胞内能量水平传感器的腺苷酸活化蛋白激酶(AMPK)也是调节自噬的重要信号通路.PI3K/AKT/mTOR 信号通路活化的结果是抑制自噬,而AMPK 信号通路活化的结果是促进自噬.
2 EGFR与肿瘤
位于细胞膜表面的EGFR(HER1 或ErbB-1)是ErbB 家族中的一个受体酪氨酸激酶,该家族还包括HER2(ErbB-2)、HER3(ErbB-3)和HER4(ErbB-4).在许多不同肿瘤细胞类型中,ErbB 通路通过各种机制被过度活化,包括配体的过表达,受体的过表达或受体的组成性活化.在正常情况下,EGFR 信号由配体结合于受体胞外域而产生.这启动受体同源/异源二聚化和胞内域自磷酸化,导致受体的活化,于是细胞质内底物被磷酸化和启动多种细胞反应的信号级联,包括基因表达的改变,细胞骨架的重排,凋亡的抑制和细胞增殖的促进.而在肿瘤中,EGFR 过表达或结构的改变均可使EGFR 信号失去控制.EGFR 过表达也成为头颈癌、卵巢癌、子宫颈癌、膀胱癌和食管癌的强有力的预后指标之一.在胃癌、乳腺癌和结直肠癌中,EGFR 的表达水平估测预后的价值相对较低[2].而在非小细胞肺癌(NSCLC)中,EGFR 高表达估测预后的价值尚存争议.EGFR 的突变也在肿瘤的发生和进展中起重要作用,常决定肿瘤对EGFR抑制剂的应答,这有助于筛选合适的患者并给予个体化治疗.
3 EGFR与自噬
EGFR 不仅在细胞膜上表达从而发挥功能,还经过内吞作用进入细胞质.细胞质内EGFR 或与内含体融合进而降解和回收利用,或转移至线粒体、细胞核发挥作用.
3.1 细胞膜EGFR调节细胞自噬通路
3.1.1 EGFR-PI3K-AKT-mTOR 通路
I 型磷脂酰肌醇三激酶(PI3K-I)是一个脂类激酶,被招募到活化的EGFR 上,并发生磷酸化作用.活化的PI3K-I 磷酸化磷脂酰肌醇2 磷酸(PIP2)为磷脂酰肌醇3 磷酸(PIP3),三磷脂酰肌醇依赖的蛋白激酶-1(PDK1)将蛋白激酶B(AKT)带到PIP3 所在的质膜上并磷酸化和活化AKT.AKT 随后活化哺乳动物雷帕霉素靶蛋白复合物1(mTORC1).活化的mTORC1 磷酸化多种与自噬启动和吞噬泡形成有关的蛋白例如ULK1、ATG13、AMBRA1 和ATG14.活化的mTORC1 也磷酸化转录因子TFEB,阻止其调节溶酶体和自噬基因的表达.最终,mTORC1 活化的结果是抑制自噬[3].
3.1.2 EGFR-RAS-ERK 通路
EGFR 经自身磷酸化后,通过结合生长因子受体结合蛋白2(GRB2)来活化鸟苷酸交换因子(SOS),SOS 使肿瘤蛋白Ras-GDP 转变为激活状态的Ras-GTP,后者最终活化胞外信号相关激酶(ERK).ERK 上调Bcl-2 家族成员Noxa 来竞争性结合该家族另一成员MCL1,从而使Beclin1 从其与MCL1 的结合中游离出来并发挥促进自噬的作用[4].
3.1.3 EGFR-JAK2-STAT3 通路
活化的EGFR 激活janus 激酶2(JAK2)来磷酸化信号转导子和转录激活子3(STAT3)的第705 位酪氨酸残基[5].STAT3 在细胞内定位不同,调节自噬的机制也不同.核内STAT3 通过提高多种自噬负调控因子(例如BCL2、BCL2L1、MCL1)的表达或者下调重要的自噬相关基因(例如Beclin1 和VPS34)来抑制自噬;而胞质内STAT3 通过分离真核翻译启动子2α 激酶2(EIF2AK2)、叉形头蛋白O1(FOXO1)和叉形头蛋白O3(FOXO3)来抑制自噬;线粒体内STAT3 通过抑制ROS 的产生来抑制自噬[6].
3.2 内含体EGFR与细胞自噬
Beclin1 是一个盘曲螺旋的蛋白,参与哺乳类动物细胞的自噬,是III 型磷脂酰肌醇三激酶(PI3K-III)复合物的其中一个成分.活化的EGFR 除可通过PI3K-AKT-mTOR 通路间接经由Beclin1 参与自噬的调节外,也可直接与Beclin1 相互作用,以不依赖mTOR 的方式调节自噬.活化的EGFR 首先经内吞作用进入细胞质,再与Beclin1 结合,使Beclin1 的Y229,Y233 和Y352 位点的酪氨酸磷酸化,增强Beclin1 与自噬负调控因子(例如Bcl-2 和Rubicon)的结合并减弱Beclin1 与VPS34 的结合,从而抑制自噬[7].而Tan 等研究发现,非活化的EGFR 也可参与自噬的调节.在血清饥饿下,非活化的EGFR与癌蛋白LAPTM4B 和囊泡外亚复合物成分Sec5 在内含体上形成复合物,该EGFR 复合物结合Rubicon,使得Beclin1 从Rubicon 中游离出来,从而启动自噬[8].
3.3 细胞核内EGFR参与细胞自噬调节
胞膜上EGFR 转运至细胞核可分为如下过程.首先是EGFR 经内吞作用进入细胞质,然后在动力蛋白的推动作用下沿着微管运动至高尔基体,最后EGFR 与高尔基体融合并继续沿着微管运动至细胞核[9].电离辐射可促使胞膜上EGFR 的核内转运,核内EGFR 活化DNA 依赖的蛋白激酶(PRKDC)来修复辐射诱导的DNA 双链破坏[10].若在电离辐射处理胶质瘤细胞的基础上抑制PRKDC 的表达,将导致大量的自噬性细胞死亡[11].然而,EGFR 的核内转移参与自噬调节还有待进一步的研究.
3.4 线粒体EGFR的非激酶依赖性自噬调节
在表皮生长因子激活下,EGFR 磷酸化并与非受体型酪氨酸激酶c-Src 共同转移到线粒体.定位到线粒体的c-Src 随后磷酸化EGFR 的第845 位酪氨酸残基并允许后者结合和磷酸化线粒体编码的细胞色素C 氧化酶亚基II(CoxII).磷酸化的CoxII 活性降低,抑制线粒体产生ATP 和ROS[12].线粒体EGFR 也可促进线粒体的增殖及线粒体在细胞伪足的聚集、诱导ATP 的产生和增强细胞的运动能力,从而导致肿瘤细胞的侵袭和远处转移[13].ATP 和ROS 均是自噬调节因子:1)细胞内的AMP/ATP 比例升高能够活化AMPK信号,从而增强自噬.2)ROS 通过结合或氧化ATG4 的第81 位半胱氨酸残基来防止LC3-I 脱脂化,从而促进自噬[14].然而,线粒体EGFR 通过调节线粒体ATP 和ROS 的产生来影响自噬的机制仍需充分研究.
4 EGFR抑制剂与肿瘤耐药
目前EGFR抑制剂主要以抑制特定的EGFR 酪氨酸激酶(EGFR-TKIs)的小分子抑制剂和以中和胞外EGFR 受体的单克隆抗体为主.EGFR-TKIs 如厄洛替尼、吉非替尼等用于治疗NSCLC 已有10 多年,取得了不错的疗效.与化疗药相比,EGFR-TKIs 在带有活化突变的NSCLC 患者中显示有更高的应答率、更长的无进展生存期和更高的生活质量[15],然而,最终大多数患者会对这些药物产生不同程度的耐药.不同于NSCLC,头颈癌很少带有EGFR 活化突变.c-Src 的活化作为EGFR 旁路途径介导头颈癌对厄洛替尼耐药,但并不影响其对西妥昔单抗的敏感性[16].在国外,EGFR-TKIs 与传统化疗药被联合用于治疗晚期胰腺癌.有研究发现,带有EGFR 突变的胰腺癌患者更可从联合用药中获益[17].但也有研究得出相反的结论,该联合用药并不比单药化疗更有效[18].因而,EGFR-TKIs临床用于治疗晚期胰腺癌仍需谨慎,并且仍需多国临床试验进一步研究.尽管EGFR-TKIs对治疗胰腺癌可能有效,肿瘤的耐药问题难以避免,有报道表明胰腺癌对EGFR-TKIs耐药与PI3K/AKT/mTOR 通路的过度活化有关[19].在其它肿瘤的治疗中,由于先天性或获得性耐药,EGFR-TKIs 的疗效也并不乐观.肿瘤细胞对EGFR-TKIs 耐药的主要机制可被划分为这三类:改变EGFR 这一药物靶点、改变相关的下游信号通路、改变其它的受体酪氨酸激酶作为EGFR 阻断的补偿旁路.最常见的耐药机制是T790M 突变,它占据50%-60%的获得性耐药病例.第二个最常见的获得性耐药机制是HER2 扩增,它占据12%的病例.然后是c-Met 扩增和PIK3CA 突变,各占据5%的病例[20].
西妥昔单抗和帕尼珠单抗均是抗细胞外域EGFR 的单克隆抗体,它们阻断配体与EGFR 的结合,导致下游RAS-RAF-MEK-ERK 信号通路的抑制.已有数个临床随机试验表明这两种抗体与传统的化疗方案联合应用可延缓带有野生型克尔斯滕大鼠肉瘤病毒癌基因同源物(KRAS)的转移性结直肠癌(mCRC)患者的疾病进展,然而,大多数患者的肿瘤最终都难免因耐药问题而加快进展.Morelli 等研究发现KRAS 的获得性耐药突变见于44%带有野生型KRAS 的患者,并且在抗EGFR 治疗后带有低频率突变KRAS的患者的无进展生存期更短[21].此外,KRAS 获得性耐药突变的肿瘤细胞可大量分泌转化生长因子-α(TGF-α)作用于邻近非耐药突变细胞,TGF-α 与EGFR 结合并导致ERK 信号的持续激活,从而介导敏感细胞的耐药[22].EGFR 的糖基化与甲基化修饰均可影响肿瘤对抗EGFR 单抗的敏感性.缺乏唾液酸糖基化的EGFR-K521变体因稳定性及与西妥昔单抗的亲和力下降而介导头颈癌对西妥昔单抗耐药[23].淋巴毒素-β 与R198 和R200 位点甲基化的EGFR 相互作用,增强甲基化介导的EGFR 与配体的结合和EGFR 的二聚化,从而促进西妥昔单抗耐药的发生[24].PI3K/AKT/mTOR 通路的组成性活化也可能与抗EGFR 治疗的耐药有关.作为PI3K-I 催化亚基的PIK3CA,已被数项研究评价其突变作为预测抗EGFR 治疗耐药的潜在价值,但该预测价值仍需大型随机对照试验来证实.受体酪氨酸激酶c-MET 和ERBB2 的基因异常也与肿瘤耐药有关,它们被认为是获得性耐药的旁路机制.近些年,随着自噬和EGFR抑制剂的不断探索与研究,人们发现自噬参与EGFR抑制剂耐药的发生或影响其治疗效果.为此,揭露确切的EGFR抑制剂耐药机制以及探索有效的抗耐药治疗策略是临床的迫切需要.
5 自噬与EGFR抑制剂耐药
在维持细胞内环境稳定中发挥重要作用的自噬,在肿瘤EGFR抑制剂耐药过程中发挥着双重作用.一方面,自噬通过移除破坏的细胞成分和蛋白来帮助肿瘤在压力下生存,导致肿瘤在治疗过程产生耐药;另一方面,自噬加强细胞凋亡的诱导或介导自噬性细胞死亡,导致疗效的增强.
5.1 自噬与EGFR-TKIs耐药
已有数项研究证实自噬介导肿瘤细胞对EGFR-TKIs 耐药,此时抑制自噬可逆转肿瘤的耐药.Liu 等研究发现,抑制自噬可增强三阴性乳腺癌(雌激素受体、孕激素受体和HER2 表达均为阴性)细胞的凋亡,从而使肿瘤对吉非替尼的敏感性提高[26].自噬抑制剂与EGFR-TKIs 的联合使用也可因增强细胞凋亡而逆转膀胱癌细胞对EGFR-TKIs 耐药[27].此外,自噬的增强也见于对厄洛替尼耐药的NSCLC 细胞,而因抑制自噬导致的细胞凋亡可由内质网应激介导[28].有研究表明,厄洛替尼诱导的自噬并不完全依赖于Beclin1 的经典自噬,用siRNA 敲除ATG5 而非Beclin1 可显著降低肿瘤细胞的生存率,该结果暗示非经典的自噬也可能参与肿瘤对EGFR-TKIs 耐药[29].由于肿瘤细胞糖酵解活性强,糖酵解抑制剂2-脱氧-D-葡萄糖(2DG)可增强厄洛替尼的体外细胞毒性作用,然而在体内却相反,2DG 可通过内质网应激介导的自噬水平的提高来减弱厄洛替尼的抗肿瘤作用[30].肿瘤获得性耐药的发生是复杂的,许多报告表明P-糖蛋白(MDR1)的过表达和自噬的上调是主要原因.装载有吉非替尼和氯喹且共轭结合了抗MDR1 单克隆抗体的壳聚糖纳米颗粒不仅可通过氯喹的作用抑制自噬,还可通过抗MDR1 单克隆抗体的作用提高吉非替尼进入细胞内的浓度,最终诱导显著的细胞凋亡.此外还发现,氯喹抑制自噬的同时,MDR1 的表达显著减少,暗示自噬可能调节MDR1 蛋白在获得性耐药的作用[31].
然而,另一些研究却显示自噬抑制介导肿瘤细胞对EGFR-TKIs 耐药,此时联合使用自噬诱导剂和EGFR-TKIs 可以提高疗效.植物精油成分日扁柏素可诱导细胞DNA 破坏和自噬来逆转肺腺癌细胞对吉非替尼耐药,若用3-MA 抑制自噬则使日扁柏素的抗肿瘤作用减弱[32].另外,其它一些具有抗肿瘤活性的药物也具有诱导自噬的作用,例如酪蛋白激酶2(CK2)抑制剂和组蛋白去乙酰化酶抑制剂.CK2 抑制剂CX-4945 能诱导吉非替尼/厄洛替尼耐药的NSCLC 细胞的自噬并可使EGFR 从细胞膜转移到自噬体中,最终导致胞膜EGFR 的减少.在克服T790M 介导的EGFR-TKIs 耐药中,CX-4945 诱导的自噬可能与EGFR-TKIs 协同抑制EGFR 信号[33].组蛋白去乙酰化酶抑制剂SAHA 和EGFR-TKIs 的联合应用能增强T790M 突变的NSCLC 细胞的自噬和凋亡,而抑制细胞自噬将显著减少联合治疗诱导的细胞凋亡.另外,该联合治疗也诱导不依赖于半胱氨酸蛋白酶(Caspase)的自噬性细胞死亡[34].
5.2 自噬与抗EGFR单克隆抗体耐药
EGFR 靶向抗体除可结合于EGFR 胞外域,阻断配体结合和EGFR 的活化,还能够介导EGFR 的内化.西妥昔单抗能够促使胞膜表面的EGFR 转移到内质网和细胞核内,而EGFR 的核内定位与肿瘤抗EGFR 治疗后的获得性耐药有关.此外,西妥昔单抗还可激发多种肿瘤细胞系的保护性自噬以介导耐药,例如,Li 等发现西妥昔单抗能够通过抑制PI3K-I/AKT/mTOR 信号通路以及增强Vps34/Beclin1 信号通路来诱导肿瘤细胞自噬以应对凋亡压力[35].自噬的诱导也见于头颈癌在抗EGFR 治疗后,通过新型自噬抑制剂spautin-1 高效且特异性地促进Vps34-Beclin1 复合物的降解能够增强抗EGFR 单抗抑制肿瘤细胞生长的作用[36].相反地,自噬也有对肿瘤不利的一面.有研究表明,西妥昔单抗与环氧合酶-2(COX-2)抑制剂联合应用能够导致抗EGFR 单抗天然耐药的结直肠癌细胞的自噬性细胞死亡[37].过度的自噬可造成肿瘤细胞死亡,因而肿瘤可能通过某些机制抵消抗EGFR 单抗诱导的过度自噬来介导耐药.自噬不仅与细胞内多条控制细胞增殖或凋亡的分子通路有关联,也与EGFR 的亚细胞定位存在一定的联系,靶向自噬抑制EGFR 的内化,阻断EGFR 进入细胞核和阻碍EGFR 的核内作用(例如DNA 修复机制),也可能是逆转肿瘤抗EGFR 单抗耐药的可行策略.
6 总结与展望
EGFR抑制剂由于其选择性高,疗效独特和毒性较小等特点,为治疗癌症开辟了一条新途径.然而,这些药物终将面临肿瘤耐药的问题.近些年来越来越多的研究证明,细胞自噬与肿瘤EGFR抑制剂耐药有关.一些研究证明,自噬的增强可介导肿瘤EGFR抑制剂耐药,同时抑制自噬和EGFR 可协同诱导肿瘤细胞的死亡.另一些研究则证明,自噬的抑制与肿瘤EGFR抑制剂耐药有关,增强自噬可诱导耐药细胞的凋亡或自噬性细胞死亡.尽管抑制自噬介导肿瘤对EGFR抑制剂耐药的相关研究较少,但却不容忽视.因此,如何正确联合应用自噬调节药与EGFR抑制剂,对于克服肿瘤耐药是至关重要的.
未来的研究应该关注这几方面:(1)寻找有效和明确的指标来指导自噬调节药与EGFR抑制剂联合应用的时机;(2)研究EGFR 甲基化与糖基化修饰在肿瘤EGFR抑制剂耐药中的作用;(3)研究自噬在EGFR抑制作用中的具体机制以及自噬在EGFR 亚细胞定位的作用;(4)自噬在P-糖蛋白介导获得性耐药过程的机制以及自噬在调节表观遗传修饰的作用,也是值得研究的方向;(5)考虑到体内组织中的肿瘤通常处于缺氧环境,自噬的研究建立在肿瘤缺氧模型上是有着重要意义的;(6)肿瘤EGFR抑制剂耐药机制也需进一步的探索.
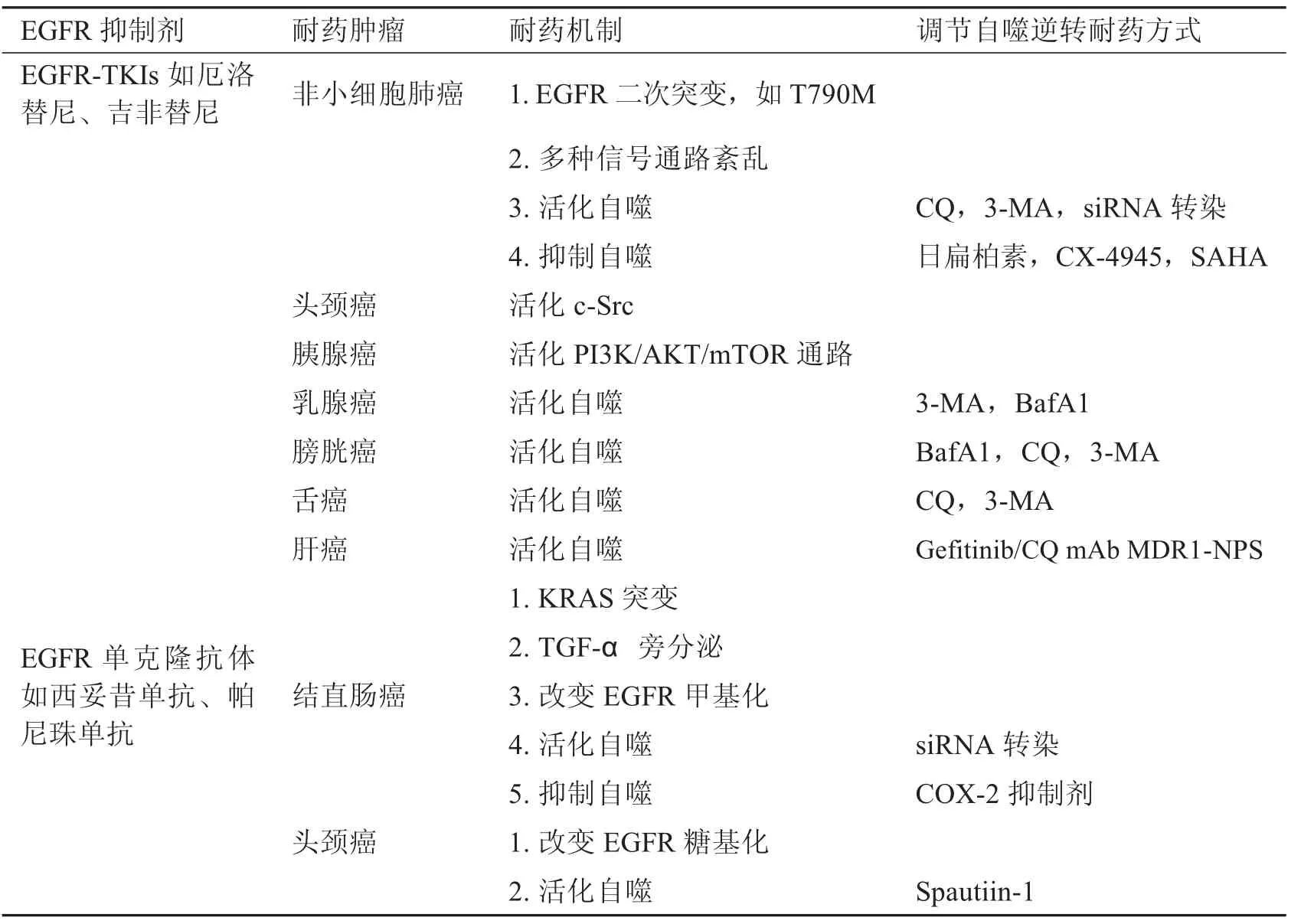
表1 肿瘤EGFR抑制剂耐药机制及调节自噬逆转耐药方式
致谢感谢蓝展鹏、周珂和戴泽宇对本文提出宝贵意见和修改!
