MoO3/Si界面区钼掺杂非晶氧化硅层形成的第一性原理研究*
2019-06-04陈东运高明李拥华徐飞赵磊马忠权
陈东运 高明 李拥华 徐飞 赵磊 马忠权
(上海大学理学院物理系,索朗光伏材料与器件R&D联合实验室,上海 200444)
1 引 言
近几年来,过渡金属氧化物由于其较高的功函数和合适的禁带宽度而作为载流子选择性接触材料,被广泛应用于光伏器件中[1-4].以MoO3为例,3.0 eV的禁带宽度使得绝大部分可见光透过,降低了寄生光吸收;6.7 eV的高功函数则可以有效地提取空穴,实现载流子的选择性接触和高效输运[2].2014年,Battaglia等[3]采用热蒸发技术在n-Si衬底上沉积MoO3薄膜,使得MoO3/n-Si结构的光伏器件的转换效率达到14.3%;随后,为了进一步提高开路电压和填充因子,Battaglia等[4]又将本征氢化非晶硅(a-Si:H(i))引入到MoO3薄膜和n-Si衬底之间,使得MoO3/a-Si:H(i)/n-Si太阳能电池的光电转换效率提升到18.8%;2015年,Geissbühler等[5]通过优化电沉积铜电极的方式,使得MoO3/a-Si:H(i)/n-Si太阳能电池的转换效率达到22.5%.然而,考虑到本征氢化非晶硅的寄生吸收和工艺窗口狭窄的限制,其他可替代的超薄钝化层也被研究者所关注.2016年,Gerling等[6]采用热蒸发的方式在n-Si表面沉积过渡金属氧化物(MoO3,WO3和V2O5)时发现,在过渡金属氧化物和硅衬底之间自然氧化形成了一层非晶氧化硅层,起到了钝化硅表面悬挂键的作用.同时,在我们先前的实验中,通过真空热蒸发的方式直接在n-Si表面沉积了8 nm的MoO3薄膜,使得Ag/ITO/MoOx/n-Si/SiOx/poly-Si(n+)/Al太阳能电池效率达到16.7%[7].通过高分辨率透射电子显微镜(high resolution transmission electron microscope,HR-TEM)、X射线光电子能谱(X-ray photoelectron spectroscopy,XPS)和X射线能量色散光谱(X-ray energy dispersive spectrum,EDS)观察,在MoO3薄膜和n-Si衬底界面处会存在一层超薄缓冲物质,厚度在3.5—4.0 nm之间,主要由Si,O和少量Mo原子组成.光伏器件良好的填充因子(72.9%)和较高的短路电流(38.2 mA/cm2),以及第一性原理计算的结果表明,钼掺杂非晶氧化硅层具有钝化接触和量子隧穿的作用,该缓冲层的存在对MoO3/Si太阳能电池的性能产生重要影响[7,8].Gerling等[6]通过计算吉布斯自由能揭示了MoO3和Si在常温下可以发生化学反应,生成SiO2和Mo,解释了氧化硅生成的可能性.然而,由于实验观察手段有限,MoO3在Si表面反应并生成钼掺杂的非晶氧化硅层的微观过程尚不明确.此外,从原子尺度深入了解钼掺杂非晶氧化硅层的形成过程和为优化实验中MoO3/Si界面提供指导,借助材料结构的数值计算方法,研究MoO3在Si(100)面上的化学反应和原子扩散具有重要的意义.
本文利用第一性原理计算方法,采用从头算分子动力学模拟了MoO3/Si界面区原子间的相互扩散、反应;研究了MoO3沉积在Si(100)表面初始时的吸附模型;并通过对相关形成能的计算(氧空位和硅、钼原子的替位掺杂),分析了Si,O和Mo三种元素的扩散情况,探讨了钼掺杂非晶氧化硅的形成机理,有利于实验中优化界面,获得更好的界面钝化效果,从而提高光伏电池的转换效率.
2 计算方法
本文的计算是利用基于密度泛函理论(density functional theory,DFT)的维也纳从头计算软件包(Viennaabinitiosimulation package,VASP)[9],选取广义梯度近似(generalized gradient approximation)下的Perdew-Burke-Eenzerhof泛函为体系交换关联能,且采用投影缀加波赝势(projector augmented wave)描述电子-离子之间的相互作用[10,11].平面波截断能选取530 eV,K点网络选择Monkhorst-Pack方法.在进行结构优化时,自洽收敛标准为:原子总能偏差小于10—4eV,且原子间的Hellmann-Feynman力偏差小于0.02 eV/Å.考虑到Mo为过渡金属,为了修正d轨道电子的强关联作用,我们分别设置了库仑作用参数U= 6.3 eV和交换关联参数J= 0 eV[12].
3 结果与讨论
3.1 钼掺杂非晶氧化硅层的分子动力学模拟
为了模拟MoO3/Si界面反应及钼掺杂非晶氧化硅层的形貌特征,构造了如图1(a)所示的周期性结构模型,该模型包含152个原子(80个Si,18个Mo,54个O),结构尺寸为10.84×11.20×28.58 Å,Si和MoO3分别选择(100)面和(010)面.为了表现出硅的块体性质,将模型中的最上和最下两层固定,其余原子在模拟中可自由移动.从头算分子动力学模拟采用正则系综[13],考虑到MoO3的沸点为1155 ℃,因此温度控制在1500 K,步长为2 fs,模拟时间为6 ps,布里渊区K点选取为4×4×1.
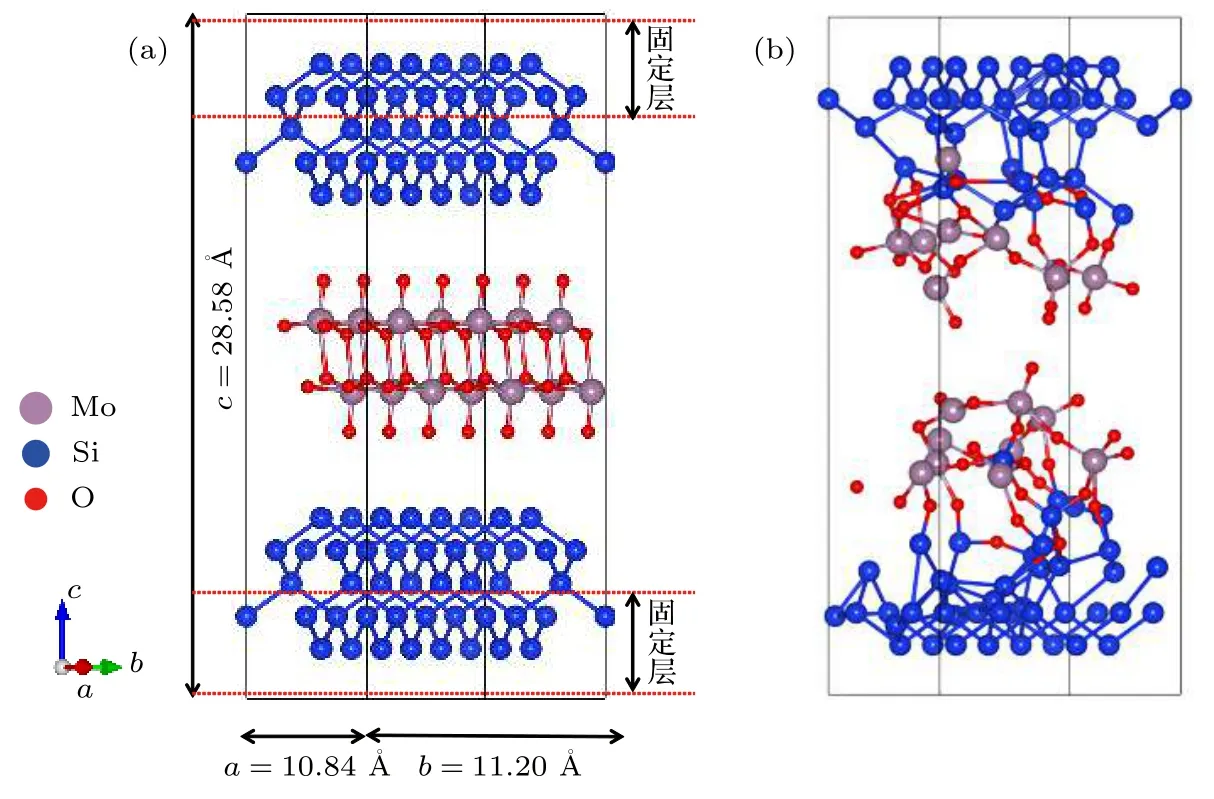
图1 MoO3(010)/Si(100)分子动力学模型 (a)扩散反应前;(b)扩散反应后;灰色球、蓝色球、红色球分别代表钼原子、硅原子和氧原子Fig.1.The structure model of MoO3(010)/Si(100)interface:(a)Before the ab initio molecular dynamics;(b)after the ab initio molecular dynamics.The grey,blue and red balls stand for Mo atoms,Si atoms,and O atoms,respectively.
分子动力学模拟结果如图1(b)所示,在MoO3/Si界面形成了一层缓冲物质.该物质由Si,O,Mo三种原子组成,这与HR-TEM,EDS和XPS实验测量获得的数据一致[8].其中,Si与O成键,构成Si—O键或Si—O—Si键,形成非晶氧化硅层;少量的Mo原子进入非晶氧化硅层中,构成Si—O—Mo键,最终构成了钼掺杂的非晶氧化硅层.
3.2 MoO3在Si(100)面上的吸附模型
MoO3薄膜的形成过程是历经了气化后的原子、分子的吸附、扩散、成团形岛并取向生长,最终形成薄膜,而了解沉积初始时的吸附模型及化学反应,有助于深入理解缓冲层的形成过程.由于MoO3粉末在蒸发过程中存在多种团簇成分[14],这里仅以MoO3分子为例.结构优化后的MoO3分子结构在图2(a)中显示,其中三个Mo—O键长均为1.73 Å,三个O—Mo—O键角分别为107.73°,107.74°和107.77°,形成四面体结构,与Oliveira等[15]的计算结果符合得很好.另外,洁净的硅表面由于存在悬挂键,为了保持稳定,通常会发生重构现象.我们选取Si(100)表面,真空层厚度设置为15 Å,采用分子动力学模拟了Si(100)表面的重构,获得经过表面重构后的Si(100)结构,如图2(a)所示.由该图可知,在硅表面发生了重构现象,表面的硅原子两两成键,形成二聚体,成键的两个原子高低不同,且前后形成高低交替的皱折结构[16].在蒸发过程中,MoO3分子可能吸附在Si(100)面的不同位置,因此用吸附能来衡量不同位置的稳定性.在Si(100)表面考虑了七个不同的位置作为初始的吸附位置[17],图2(a)中用不同颜色来表示不同的初始位点,其中包括位点1(黄)、2(橙)、3(绿)、4(蓝)、5(粉红)、6(红)、7(紫).我们采用了吸附能的定义[18]:

图2 MoO3在Si(100)不同吸附位点的结构示意图 (a)MoO3分子结构及重构后Si(100)表面形貌;(b)-(h)MoO3在吸附位点1-7时优化后的吸附模型;(i)最佳吸附位点7的差分电荷密度(黄色和绿色表示得失电子)Fig.2.Adsorption configurations of MoO3on Si (100)surface:(a)The optimized geometries of MoO3molecule and reconstructed Si(100);(b)-(h)the adsorption configurations of MoO3adsorbed on the different adsorption sites of Si (100)surface;(i)the difference charge density of MoO3on the best adsorption site 7 of Si (100).

其中,Eab表示吸附能,ESi/MoO3表示MoO3吸附在Si(100)后体系的总能量,ESi和EMoO3则分别为Si(100)表面和MoO3分子的总能量.
MoO3沉积在Si(100)表面的吸附模型和吸附能如图2和表1所示.由图2可知,沉积后MoO3和Si表面结构都发生了不同程度的变化,形成了Mo—Si键或Si—O键.由表1可知,MoO3吸附在位点7时最为稳定,吸附能为—5.36 eV,且在吸附位点1时最不稳定,吸附能仅为—2.36 eV.再者,结合图2和表1可知,吸附位点2—7的吸附能都比吸附位点1要小,这说明相比于Si—Mo成键,Si和O成键更加稳定.所以,在沉积过程中,O原子会优先和Si原子发生反应,生成氧化硅;吸附位点3—7的吸附能又都比吸附位点2要小,这说明MoO3在吸附时,更倾向于两个O原子同时与两个Si成键,形成两个Si—O键.为了详细了解吸附后结构参数的变化和电子的转移情况,我们选择了最佳吸附位点7进行分析,结果如图2(h)—(i)和表2所示.Si原子和O原子成键,键长为1.68 Å.MoO3的键长和键角也都发生了明显的变化,Mo—OI,Mo—O∏的键长分别从1.73 Å扩大到1.95 Å和1.94 Å.MoO3吸附时会伴随着原子间电荷的重新分配,通过Bader电荷分析可知[19],电荷转移主要发生在SiI,Si∏,OI和O∏之间,Si失去电子,O得到电子.为了说明存在电荷的转移,我们用体系的差分电荷密度[20](Δρ=ρMoO3/Si(100)-ρSi(100)-ρMoO3(ρMoO3/Si(100),ρSi(100),ρMoO3分别表示吸附后MoO3/Si(100)整个体系、单独Si(100)体系、单个MoO3的电荷密度))来表示这种转移,其结果表示在图2(i)中.

表1 MoO3在Si(100)不同吸附位点的吸附能Table 1.The adsorption energy of MoO3on Si (100).
3.3 钼掺杂非晶氧化硅的形成机理
由前面的计算结果可知,在沉积初期,MoO3的O原子和晶硅衬底表面的Si原子成键更加稳定,钝化了Si表面的悬挂键.同时,当缓冲层厚度达到3.5—4.0 nm时,薄膜中存在着Mo元素掺杂[8],这必然伴随着Mo,O原子的移动.因此,下面我们将从形成能的角度探究钼掺杂非晶氧化硅层的形成原因.
首先,考虑了中性条件下α-MoO3和α-SiO2中氧空位的形成能,借此分析O原子的移动,其中α-MoO3和α-SiO2晶胞结构如图3所示.在MoO3中,O原子存在三种不等价的位置,分别标记为O1,O2和O3[21].为了排除晶格大小带来形成能的误差,我们选择了3×1×3的α-MoO3(共144个原子)和2×2×2的α-SiO2(共72个原子).氧空位形成能定义为[22]

表2 吸附前后体系7结构参数变化及Bader电荷Table 2.The structure parameters and Bader charge of MoO3adsorbed on the adsorption site 7 of Si (100)surface.
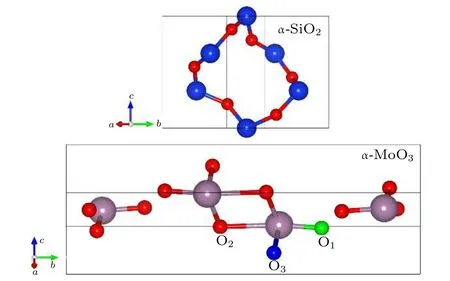
图3 α-SiO2和α-MoO3晶胞结构Fig.3.The framework of α-SiO2and α-MoO3unit cells.

其中,X代表α-MoO3或者α-SiO2;表示α-MoO3或α-SiO2中氧空位的形成能;表示存在一个氧空位的体系的总能量;表示完整晶体体系的总能量;µ(O)表示O的化学势,由一个孤立氧分子总能量的一半计算得到.
由计算可知,α-SiO2中氧空位的形成能为5.11 eV[22],而α-MoO3中三种氧空位的形成能分别为0.96,1.96和3.19 eV[21].两者对比可知,α-MoO3中氧空位的形成能均小于α-SiO2中氧空位的形成能,说明α-MoO3中的氧空位更容易形成.在MoO3中较低的氧空位形成能意味着O原子容易摆脱Mo—O键的束缚,迁移进入Si一侧,从而和Si反应,形成3.5—4.0 nm的非晶氧化硅层.形成能的计算也表明,若在MoO3/Si界面处存在不饱和氧化硅层时,氧原子将从MoO3向不饱和氧化硅层移动,填补氧化硅层中的氧空位,Liu等[23]的实验则证实了我们的推测,他们在对样品进行后退火时,发现O原子向非晶氧化硅一侧移动,使得不饱和氧化硅(SiOx)向SiO2转变.
在考虑了O原子的扩散后,进一步研究Mo原子是如何掺杂进入非晶氧化硅层的.这里我们考虑了Mo,Si原子相互替位的形成能,即在α-MoO3中用一个Si原子替位一个Mo原子(SiMo)或者在α-SiO2中用一个Mo原子替位一个Si原子(MoSi).SiMo或者MoSi的形成能分别定义为[22]:

或者

从形成能的计算结果,如图4(a)中,可以看到在富Si条件下(µ(Si(bulk))),可能出现Si原子替位MoO3中的Mo原子,然而随着Mo的富集,形成能越来越大,替位越来越困难.此外,富氧条件下(µ(Si(gas))),Si原子替位Mo原子几乎不可能发生.结合前面的吸附能计算可知,MoO3沉积在Si(100)表面,O原子与Si原子成键更加稳定,使得表面优先形成氧化硅.而随后的发展使得Si原子替位Mo原子进入MoO3薄膜非常困难.再如图4(b)中,在富氧条件下(µ(Si(gas))),容易发生Mo原子替位SiO2中的Si原子,且随着Mo的富集,替位将越来越容易.然而在富Si条件下(µ(Si(bulk))),Mo的替位则变得相对困难,这意味着Mo原子很难替位晶体硅中Si原子,而容易替位氧化硅中的Si原子,这使得Mo容易掺杂进入氧化硅中,并最终形成钼掺杂的非晶氧化硅层.

图4 不同生长条件下替位杂质形成能 (a)Si替位Mo;(b)Mo替位Si;黑线表示晶体硅的化学势,红线表示富氧条件下硅的化学势Fig.4.The formation energy for the two substitutional defects:(a)Si in place of a Mo in MoO3;(b)Mo in place of a Si in SiO2.The black curves stand for the bulk Si chemical potential and the red curves stand for the chemical potential for Si in the SiO2under an oxygen-rich environment.
综上所述,根据吸附和缺陷形成能的计算可知,在沉积过程中,Mo,O,Si三种原子的迁移情况如下:当MoO3沉积在Si表面时,O优先和Si成键,MoO3中的O原子从MoO3一侧向晶体Si扩散,形成氧化硅层;随后失去O原子的Mo原子也向非晶氧化硅一侧扩散,替位氧化硅中的Si原子,从而最终形成含有钼掺杂的非晶氧化硅层.
4 结 论
本文利用基于密度泛函理论的第一性原理计算,并结合实验数据,从分子动力学、吸附能、缺陷形成能多个角度,构建了MoO3在Si(100)面的吸附模型,探究了钼掺杂非晶氧化硅层的形成原因.热蒸发时,MoO3以O原子和Si原子成键的方式吸附在Si衬底表面上,且在吸附位点7处吸附最为稳定,吸附能为-5.36 eV.扩散过程中,由于SiO2中O空位的形成能为5.11 eV,而MoO3中的三种O空位形成能分别为0.96,1.96和3.19 eV,使得O原子容易从MoO3中迁移至Si衬底一侧,从而形成氧化硅层,起到了界面钝化的作用;再者由于低的替位形成能,使得部分Mo原子掺杂进入氧化硅中,最终形成了具有钝化接触和量子隧穿功能的α-SiOx(Mo)层.本文加深了对于MoO3沉积在Si表面的反应过程及原子扩散情况的理解,为实验优化MoO3/n-Si界面提供了理论依据.
感谢上海大学高性能计算中心提供的计算资源.
