都梁方中挥发油β-环糊精包合物的制备
2019-06-01韦小翠杨书婷朱宇超颜媛媛程建明柳春娣
韦小翠, 杨书婷, 张 焱, 朱宇超, 颜媛媛, 程建明, 柳春娣
(1. 南京中医药大学药学院, 江苏省中药功效物质重点实验室, 江苏 南京 210023; 2. 南京中医药大学附属张家港医院, 江苏 张家港 215600)
都梁方最早记载于宋代《是斋百一选方》, 为王璆博采民间验方而来, 原方由白芷研末加炼蜜为丸而成, 后人在此基础上加川芎炼蜜为丸(1983年版《北京市药品标准》 ), 即“都梁丸”[1], 现收载于2015 年版《中国药典》, 用于治疗风寒之邪上犯头部、 清阳之气受阻、 气血凝滞引起的头痛[2-4]。 方中白芷、 川芎均含有挥发油, 前者具有明显的镇痛、 镇静、 抗过敏作用[5], 而后者有着解热、 镇痛镇静、 改善血流变、 保护神经细胞、 抗炎、 降压等活性[6], 两者药效与全方基本相符,即是有效成分, 但该类成分具有挥发性, 而且在氧、 光、 热的作用下极易分解变质, 有不易贮存、生物利用度低等缺点。 因此, 本实验制备都梁方中挥发油β-环糊精包合物, 使液体药物粉末化, 增加挥发油稳定性, 以便进一步制成不同剂型, 为今后相关研究提供基础。
1 材料
1.1 仪器 DF-101S 集热式恒温加热磁力搅拌器(巩义市予华仪器有限责任公司); 高速分散仪(宁波新芝生物科技股份有限公司); KQ-500DE 数控超声波清洗器(昆山市超声仪器有限公司);HH-2 数显恒温水浴锅(上海维诚仪器有限公司);Biomate 3S 紫外可见分光光度计、 Nicolet is5 傅立叶变换红外光谱仪[赛默飞世尔科技(中国) 有限公司]。
1.2 试药 挥发油 (自制)。 β-环糊精 (批号Z28F8Y30171 )、 羟 丙 基-β-环 糊 精 ( 批 号Y15S8C43689) (上海源叶生物科技有限公司);水为蒸馏水。
2 方法与结果
2.1 挥发油提取 采用水蒸气蒸馏法。 按照前期预实验确定的最优工艺, 即粉碎12 ~24 目, 加10倍量水浸泡0.5 h 后提取6 h, 得淡黄色油状液体,加入无水硫酸钠脱水, 5 000 r/min 离心5 min 后,移取上层液体, 即得。
2.2 含油量测定(紫外分光光度法)[7]
2.2.1 方法学考察
2.2.1.1 检测波长筛选 精密称取挥发油约0.5 g, 置于100 mL 量瓶中, 无水乙醇定容至刻度。 另外精密称取β-环糊精、 包合物各约0.5 g,置于50 mL 锥形瓶中, 精密加入20 mL 无水乙醇,超声30 min 后过滤, 续滤液稀释至一定质量浓度后测定, 以无水乙醇为空白, 在200 ~400 nm 波长范围内进行全波长扫描。 结果, 挥发油、 包合物提取液在320 nm 波长处均有最大吸收, 吸收曲线相似, 而β-环糊精提取液在该该处没有吸收, 不干扰测定, 故选择检测波长为320 nm。
2.2.1.2 线性关系考察 精密移取挥发油0.5 g,置于100 mL 量瓶中, 无水乙醇稀释至刻度, 精密移取1 mL 置于50 mL 量瓶中, 无水乙醇定容, 作为对照溶液, 再精密吸取1.5、 2.0、 2.5、 3.0、3.5、 4.0、 4.5 mL 置于10 mL 量瓶中, 无水乙醇定容至刻度, 摇匀, 在320 nm 波长处测定吸光度。以吸光度(A) 对挥发油质量浓度(X) 进行回归, 得方程为A=16 829X+0.001 96 (r=0.999 5),在1.5×10-5~4.5×10-5g/mL 范围内呈良好的线性关系。
2.2.1.3 精密度试验 精密吸取对照溶液适量,按“2.2.1.2” 项下方法测定吸光度, 重复5 次,测得其RSD 为0.33%, 表明仪器精密度良好。
2.2.1.4 重复性试验 精密称取包合物5 份, 每份约0.5 g, 精密加入20 mL 60% 乙醇, 超声30 min后过滤, 精密吸取适量续滤液, 60%乙醇稀释至适当质量浓度, 在320 nm 波长处测定吸光度,测得其RSD 为0.41%, 表明该方法重复性较好。
2.2.1.5 加样回收率试验 取含油量已知的包合物9 份, 分为3 组, 分别按50%、 100%、 150%水平加入挥发油, 测定含油量。 结果, 平均加样回收率为97.5%, RSD 为0.70%。
2.2.2 测定方法 精密称取包合物约0.5 g, 精密加入20 mL 60%乙醇, 超声30 min 后过滤, 精密吸取续滤液适量, 60% 乙醇稀释约1 000 倍, 于320 nm 波长处测定吸光度, 代入“2.2.1.2” 项下回归方程测得挥发油质量浓度, 计算含油量及包合物收率、 含油率、 包合率, 公式如下。
含油量= (挥发油质量浓度×溶液稀释倍数×溶液体积) /挥发油质量×包合物质量
包合物收率= [包合物实际质量/(β-环糊精加入量+投油量) ] ×100%
含油率= (包合物含油量/包合物总质量) ×100%
包合率= (包合物实际含油量/投油量) ×100%
2.2.3 综合评价 包合率是评价包合效果的主要指标, 包合率越高, 表明挥发油包合效果越好, 可有效改善挥发油稳定性, 有利于制剂稳定, 故将其权重系数确定为0.6[8]; 包合物收率在实际生产中具有重要意义, 但辅料加入过多时会增加成本, 故将其作为次要考察指标, 权重系数确定为0.2; 含油率是评价包合物的重要指标之一, 在控制制剂质量方面有重要意义, 故将其权重系数确定为0.2[9-11], 综合评分=包合率×0.6 +包合物收率×0.2+含油率×0.2。
2.3 包合物制备
2.3.1 包合材料
2.3.1.1 β-环糊精包合物[12]称取β-环糊精2 g,置于100 mL 锥形瓶中, 加入40 mL 蒸馏水, 加热溶解, 制备饱和水溶液[13], 转移至恒温磁力搅拌器中, 搅拌下逐滴加入0.5 mL 挥发油(等量无水乙醇溶解), 转速300 r/min, 温度40 ℃, 包合时间2 h, 待其冷却至室温后, 于4 ℃冰箱中冷藏24 h, 布氏漏斗抽滤, 依次用10 mL 蒸馏水、 无水乙醇(约30 mL, 10 mL/次) 洗涤, 置于45 ℃烘箱中干燥4 h, 即得。
2.3.1.2 羟丙基-β-环糊精包合物 称取羟丙基-β-环糊精2 g, 置于100 mL 锥形瓶中, 加入40 mL蒸馏水, 加热溶解, 制备饱和水溶液, 转移至恒温磁力搅拌器中, 搅拌下逐滴加入0.5 mL 挥发油(等 量 无 水 乙 醇 溶 解), 转 速300 r/min, 温 度40 ℃, 包合时间2 h, 待其冷却至室温后, 分次加入10 mL 石油醚摇匀, 5 000 r/min 离心10 min,移去上层液体, 除去未包合的挥发油, 下层液体冷冻干燥, 即得。
2.3.1.3 包合效果比较 精密称取2 种包合物各约0.5 g, 按“2.2” 项下方法测定含油量, 计算包合物收率、 包合率、 含油率, 结果见表1。 由表可知, 以β-环糊精为包合材料时综合评分较高,其包合率、 含油率均高于羟丙基-β-环糊精, 其原因可能是后处理方式不同所致; 由于羟丙基-β-环糊精水溶性很好, 在与β-环糊精相同的处理方式(即于4 ℃冰箱中冷藏24 h) 下并未出现包合物与溶液分层的现象, 故无法进行抽滤, 而采用冷冻干燥方式制备, 但该方法耗时长, 生产成本较高, 综合考虑, 选择β-环糊精作为包合材料。
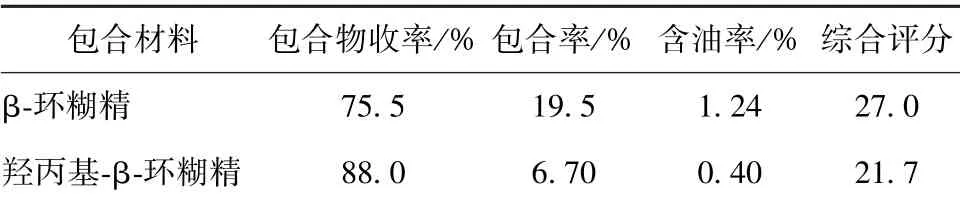
表1 不同包合材料包合效果比较Tab.1 Comparison of inclusion effects of different inclusion materials
2.3.2 制备方法[14]
2.3.2.1 磁力搅拌法 同“2.3.1.1” 项。
2.3.2.2 高速分散法 称取β-环糊精2 g, 置于100 mL 锥形瓶中, 加入40 mL 蒸馏水, 加热溶解,制备饱和水溶液, 转移至40 ℃高速分散仪中, 逐滴加入0.5 mL 挥发油(等量无水乙醇溶解), 在相应温度下高速分散后, 取出冷却至室温, 余下步骤同“2.3.1.1” 项。
2.3.2.3 超声法 称取β-环糊精2 g, 置于100 mL锥形瓶中, 加入40 mL 蒸馏水, 加热溶解,制备饱和水溶液, 转移至40 ℃超声仪中, 逐滴加入0.5 mL 挥发油(等量无水乙醇溶解), 40 ℃下超声1 h 后, 取出冷却至室温, 余下步骤同“2.3.1.1” 项。
2.3.2.4 研磨法 称取β-环糊精2 g, 置于研钵中, 加入40 mL 蒸馏水, 逐滴加入0.5 mL 挥发油(等量无水乙醇溶解), 研磨40 min 后转移至锥形瓶中, 余下步骤同“2.3.1.1” 项。
2.3.2.5 包合效果比较 精密称取4 种包合物各约0.5 g, 按“2.2” 项下方法测定含油量, 计算包合物收率、 包合率、 含油率, 结果见表2。 由表可知, 磁力搅拌法、 研磨法所得包合物收率较高,而高速分散法、 超声法较低, 其原因可能是包合物在洗涤过程中部分流失, 导致粘于器壁内部者未能收集而出现误差; 高速分散法所得包合率、 含油率最高, 其他3 种方法相差不大, 综合考虑, 选择高速分散法作为包合方法。
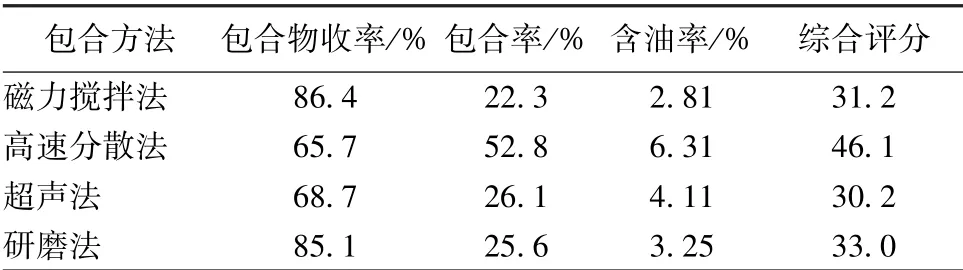
表2 不同包合方法包合效果比较Tab.2 Comparison of inclusion effects of different inclusion methods
2.3.3 工艺优化 通过预实验及查阅文献[15-18], 选择挥发油与β-环糊精比例(A)、 包合温度(B)、 分散时间(C) 作为影响因素, 包合物收率、 包合率、 含油率作为评价指标, 正交试验优化制备工艺。 因素水平见表3, 结果见表4, 方差分析见表5。
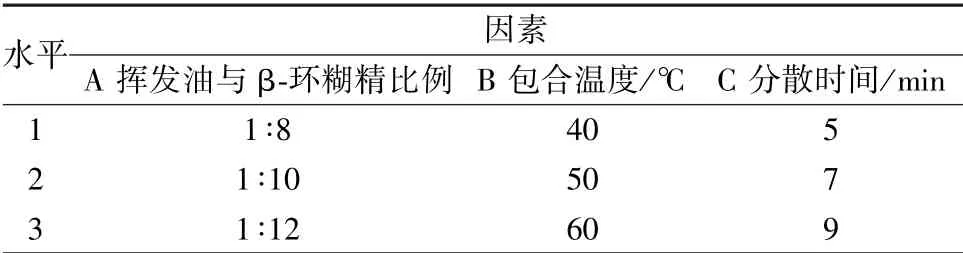
表3 因素水平Tab.3 Factors and levels
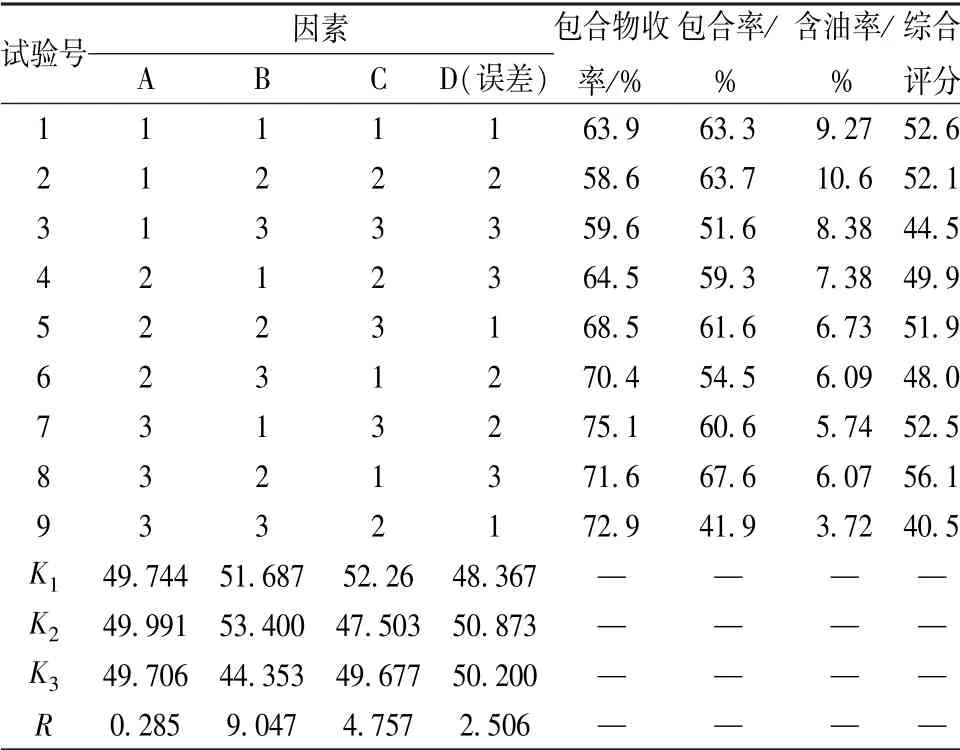
表4 试验设计及结果Tab.4 Design and results of tests
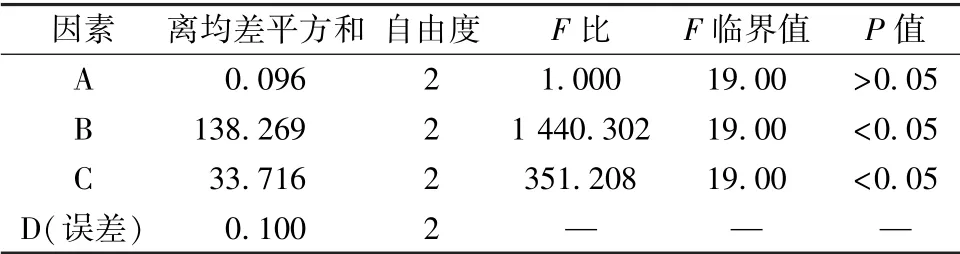
表5 方差分析Tab.5 Analysis of variance
表4 显示, 各因素影响程度依次为B>C>A;表5 显示, 因素B、 C 有显著影响(P<0.05)。 由此确定, 最优工艺为A2B2C1, 即挥发油与β-环糊精比例1 ∶10, 包合温度50 ℃, 分散时间5 min。
然后, 制备3 批包合物, 按上述优化工艺进行验证试验, 结果见表6, 可知该工艺稳定可行。
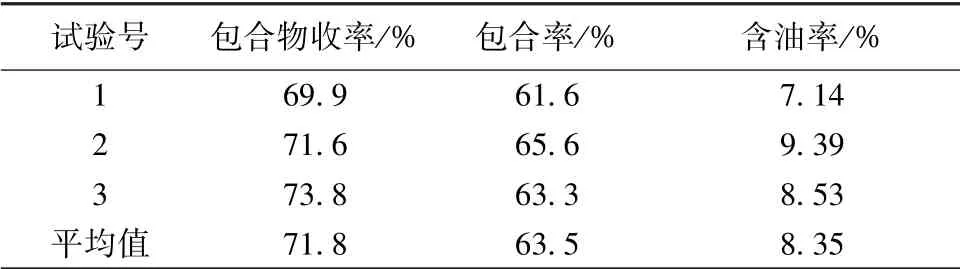
表6 验证试验结果(n=3)Tab.6 Results of verification tests (n=3)
2.4 包合物质量评价
2.4.1 显微镜法 取β-环糊精、 高速分散后的β-环糊精、 包合物少许装片, 置于载玻片上, 显微镜观察其成像, 并拍摄照片, 结果见图1。 由图可知, β-环糊精、 高速分散后的β-环糊精均为无色透明立方体状结晶, 表明外力作用并未使其形态发生改变; 包合物为黑色团状物质, 表明发生了包合, 同时β-环糊精形态变化也表明有新物相形成。
2.4.2 红外光谱法 取β-环糊精、 包合物、 物理混合物适量, 采用溴化钾压片法, 分辨率4 cm-1,扫描范围4 000~400 cm-1, 进行红外光谱测定, 结果见图2。 由图可知, 物理混合物在1 766、 1 667、706 cm-1处有较强吸收峰, 而包合物中相应波长处的吸收峰在包合后强度明显减弱或消失, 峰形与β-环糊精较接近, 表明发生了包合。

图1 样品显微图Fig.1 Microscopic images for samples
2.4.3 差示扫描量热法[19]称取β-环糊精、 物理混合物、 包合物各8 mg, 置于铝制坩埚中, 氮气体积流量40 mL/min, 升温速度10 ℃/min, 升温范围25~350 ℃, 结果见图3。 由图可知, 药物分子与β-环糊精分子的包合是焓、 熵共驱过程, 吸热峰消失、 峰型变宽、 峰转移或新峰产生表明晶格、 烙点、 沸点、 升华点等改变[20]; β-环糊精在120、 220 ℃下有明显特征峰, 前者是结晶水蒸发的吸热峰, 而后者是β-环糊精的熔融分解峰[21];物理混合物图谱与β-环糊精基本相似, 但前者含有低沸点的挥发性成分, 其吸热峰提前至106 ℃;包合物在220 ℃下未出现β-环糊精特征峰, 表明后者空腔内结晶水已被挥发油置换出来, 生成了新物相, 而且吸收峰较平缓, 热熵值减小, 可能发生了分子作用, 推测是由于包合效应的存在而使得包合物热稳定性提高。
3 讨论
本实验比较了β-环糊精、 羟丙基-β-环糊精的包合效果, 发现两者水溶性相差极大, 后处理步骤也有所不同, 其中前者所得包合物收率虽然略低,但包合率、 含油率、 综合评分高于后者, 故选择其作为包合材料。 然后, 比较了磁力搅拌法、 高速分散法、 超声法、 研磨法的包合效果, 发现参数条件相同时, 超声法、 磁力搅拌法、 研磨法有一部分挥发油会浮在液面上, 并未被包合进去, 从而造成损失; 高速分散法未发现这种现象, 而且操作简便,时间短, 在一定程度上降低了生产成本, 有利于挥发油包合工业生产, 故选择其作为制备方法。
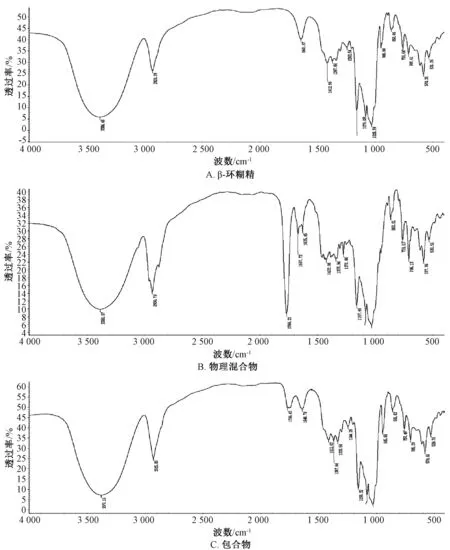
图2 样品红外光谱图Fig.2 Infrared spectra for samples
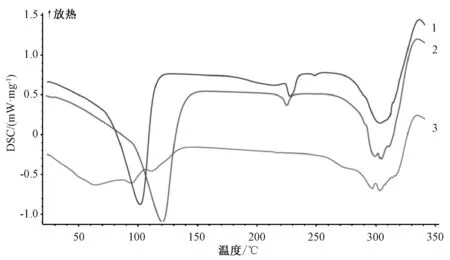
图3 样品差示扫描量热图谱Fig.3 Differential scanning calorimetry spectra for samples
在挥发油β-环糊精包合物的质量评价中, 大部分文献均采用经典的《中国药典》 方法, 即水蒸气蒸馏法进行提取测定, 但由于都梁方中川芎挥发油的密度与水接近, 极易发生乳化, 给测定造成了一定困难, 而且该方法测定时间较长, 读数小,误差较大。 当体系达到动态平衡时, 解吸附的逆向反应也将达到平衡, 不能彻底进行; 有些解吸附成分溶于沸水中, 造成包合物中挥发油提取不完全,影响其包合率测定[22], 故本实验采用紫外分光光度法, 能快速、 准确地对都梁方中挥发油β-环糊精包合物进行含油量测定。
另外, 本实验以乙醇为提取溶剂, 浸泡后超声提取包合物中挥发油, 再考察了不同体积分数乙醇对提取效果的影响, 发现60% 时最理想, 故选择60%乙醇作为提取溶剂。
