柱后衍生化-HPLC法检测硫酸新霉素的有关物质
2019-06-01张杨慧蒋孟虹赵敬丹秦峰刘浩
张杨慧 蒋孟虹 赵敬丹 秦峰,3 刘浩,*
(1 中国医药工业研究总院,上海 201203;2 上海市食品药品检验所,上海 201203;3 复旦大学化学系,上海 200433)
新霉素(neomycin)是首个被发现的2-脱氧链霉胺双取代氨基糖苷类抗生素[1]。由于硫酸新霉素的有关物质较多[2](图1),杂质(如新霉胺)一般活性较低且具有一定的毒性,会影响临床疗效,导致不良反应,因此对新霉素进行有关物质的检测是产品质量控制十分重要的环节。
新霉素及其有关物质缺乏特征的紫外吸收,中国药典2015年版[3]采用TLC法仅控制新霉胺限度。欧洲药典9.0版[2]收载的硫酸新霉素有关物质检测方法为HPLC-脉冲安培检测(PAD)法,流动相为三氟乙酸溶液,但此方法并不能将杂质LP-B与新霉素B分离,且新霉胺与杂质LP-A不能完全分离[5]。虽然Hoogmartens等[4-5]改进了欧洲药典方法,但该类分析系统价格昂贵,对操作人员的技能要求较高,不利于检测方法的普及。国内外文献报道硫酸新霉素的有关物质分析方法还有HPLC-蒸发光散射检测(ELSD)法[6-7],柱前衍生化-HPLC法[8-9],LC-MS法[10-13]等。ELSD具有通用性强、维护要求低等优点,但其要求流动相具有挥发性,这使得色谱分离系统的优化受到较大的限制。柱前衍生化法通常采用的衍生化试剂为邻苯二甲醛,茚三酮等,但该方法操作繁琐,且柱前衍生物不稳定,易发生荧光淬灭。文献报道两种LC-MS法,第一种[10]采用三氟乙酸/五氟丙酸色谱系统分离,但该流动相中的多氟羧酸会抑制质谱离子化效率。第二种[11-13]是采用HILIC柱,但该色谱柱无法将新霉素及其有关物质有效分离,仅在血浆,动物的药物残留检测中应用较多。
本文参考文献[5]采用分离能力较强的离子对HPLC分离系统,新建了硫酸新霉素有关物质检查的柱后衍生化-HPLC-荧光检测法。该方法操作简便,灵敏度高,不仅提高了色谱分离能力,同时避免了昂贵的电化学检测器的使用,有利于方法的普及,可作为新霉素有关物质检测的常规方法。本文同时将新建HPLC分离系统与电化学检测器联用,并对两种方法进行了比较。
1 仪器与材料
仪器:Agilent 1100高效液相色谱仪,Agilent 1100荧光检测器,Pickering PCX5200柱后衍生化系统,ICS-5000+型离子色谱仪(Thermo Scientific),Waters 2767/2545/SFO制备液相色谱仪。
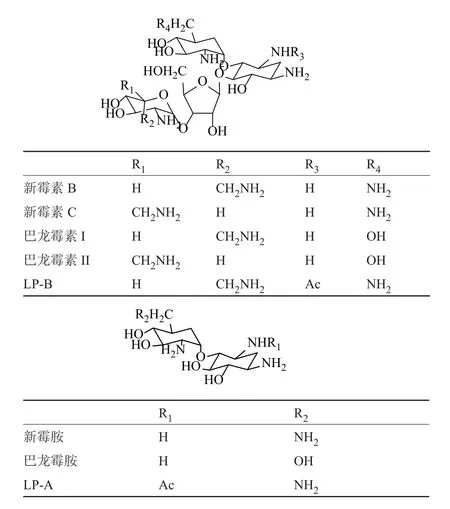
图1 新霉素及其主要杂质结构Fig.1 Structures of neomycin and its chief impurities
庚烷磺酸钠一水合物(色谱纯)由东京化成工业株式会社提供;乙腈和甲醇(色谱纯)由Merck公司提供;冰醋酸、无水硫酸钠、硼酸、氢氧化钠和2-巯基乙醇(分析纯)均由上海凌峰化学试剂有限公司提供;邻苯二甲醛(色谱纯)由北京百灵威科技有限公司提供;水为纯化水。电化学检测系统采用的无水硫酸钠由SIGMA提供,50%氢氧化钠溶液由ACROS提供。
硫酸新霉素标准品(批号:130309-200811,中国食品药品检定研究院);硫酸新霉素对照品(批号:45800,USP对照品);硫酸新霉素对照品(批号:N0400000,Batch3.7欧洲药典);弗莱菌素(新霉素B组分,华曙制药集团);新霉素C(华曙制药集团);新霉胺(批号:30411-200908,中国食品药品检定研究院);核糖霉素(批号:3489301,中国食品药品检定研究院);巴龙霉素I(paromomycin sulfate批号:6-EOD-120-1,Toronto Research Chemicals);巴龙霉胺(批号:P195340,Toronto Research Chemicals);硫酸新霉素(批号:N-9209-56、N-9301-1、N-9705-113B、N-9704-107A,上海新亚药业有限公司)。
2 方法
2.1 色谱分离系统
2.1.1 柱后衍生化-HPLC法色谱系统
色谱柱:COSMOSIL 5C18-PAQ(4.6mm×250mm,3.5μm);流动相A:pH3.4缓冲液[取庚烷磺酸钠一水合物4.35g和无水硫酸钠16g,加水溶解并稀释至1000mL,用冰醋酸调节pH值至(3.4±0.1)]-乙腈(95:5);流动相B:pH3.4缓冲液-乙腈(60:40);按表1进行线性梯度洗脱;流速:1.0mL/min;柱温30℃;进样体积:5μL。
柱后衍生系统:衍生化试剂[取邻苯二醛0.8g、甲醇300mL、2-硫基乙醇2mL和硼酸盐缓冲液(称取72.0g硼酸和43.0g氢氧化钠,加水溶解并稀释至4000mL,用1mol/L硼酸溶液或1mol/L氢氧化钠溶液调节pH值至10.4±0.1)700mL,混匀,用0.45μm滤膜滤过]的流速为0.3mL/min。
荧光检测器:激发波长:340nm,发射波长:455nm。
2.1.2 HPLC-PAD法色谱系统
同柱后衍生化法-HPLC的色谱分离条件,进样体积为25μL。
柱后碱溶液(量取超纯水974mL,氦气脱气10min后,量取50%氢氧化钠溶液26mL,混合,继续通氦气脱气2min即得);流速为0.3mL/min。
2.2 溶液的配制
2.2.1 柱后衍生化-HPLC法
系统适用性溶液:取国产硫酸新霉素对照品约39mg,置于50mL量瓶中,加水溶解并稀释至刻度,摇匀即得(0.5mg/mL)(图2A)。
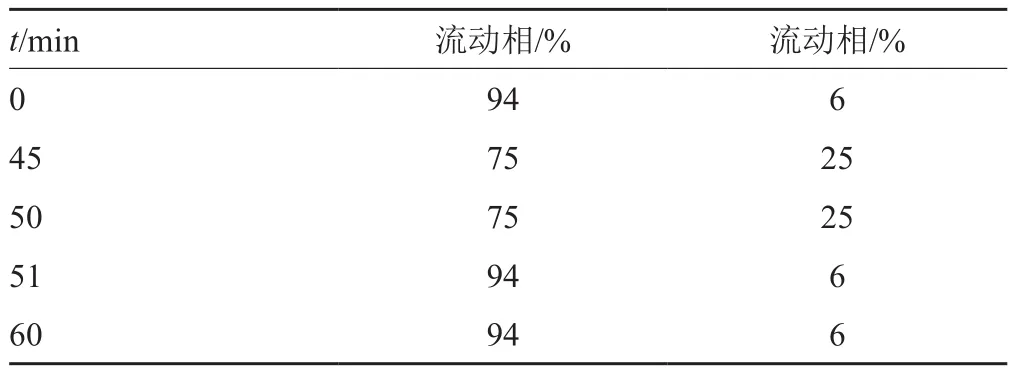
表1 梯度洗脱程序Tab.1 Gradient elution procedures
供试品溶液:取硫酸新霉素适量,加水溶解并稀释制成浓度为0.5mg/mL的溶液。
2.2.2 HPLC-PAD法
系统适用性溶液:取国产硫酸新霉素对照品约23mg,置于100mL量瓶中,加水溶解并稀释至刻度,摇匀即得(0.15mg/mL)(图2B)。
供试品溶液:取硫酸新霉素适量,加水溶解并稀释成浓度为0.15mg/mL的溶液。
3 方法学考察
3.1 专属性
取硫酸新霉素约78mg,精密称定,置于10mL量瓶中,加水溶解并稀释至刻度,摇匀,作为样品贮备液(5mg/mL);酸破坏试验:取样品贮备液1mL,加2mol/L盐酸0.5mL,混匀,室温放置18h,加2mol/L氢氧化钠溶液0.5mL中和。碱破坏试验:取样品贮备液1mL,加2mol/L氢氧化钠0.5mL,混匀,室温放置18h,加2mol/L盐酸溶液0.5mL中和。氧化破坏试验:取样品贮备液1mL,加30%双氧水1mL,混匀,室温放置1h。高温破坏试验:取样品贮备液1mL,于105℃高温放置8h。光照破坏试验:取样品贮备液1mL,于254和365nm紫外光放置18h。分别将上述破坏样品用纯水稀释制成约含新霉素0.5mg/mL的溶液。按“2.1.1”项下色谱条件进样分析。色谱图如图3所示。主峰与降解杂质峰之间均分离良好,新建方法的专属性良好。
3.2 重复性
精密称取同一批号(N-9705-113B)的硫酸新霉素6份,加水溶解并稀释制成约含新霉素0.5mg/mL的溶液,进样分析,按面积归一化法计算,新霉胺均为0.8%;核糖霉素的平均值为0.8%,RSD为0.02%(n=6);巴龙霉素I的平均值为1.45%,RSD为0.02%(n=6);杂质总量的平均值为7.6%,RSD为0.3%(n=6),该方法重复性良好。
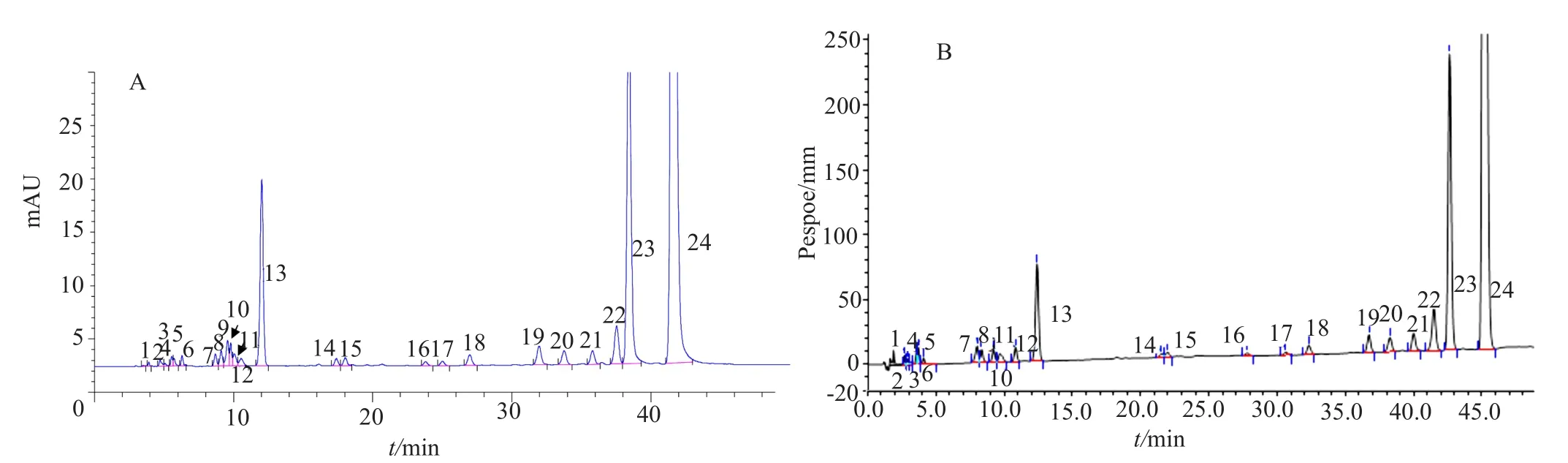
图2 系统适用性溶液典型色谱图Fig.2 HPLC chromatogram for system suitability solution
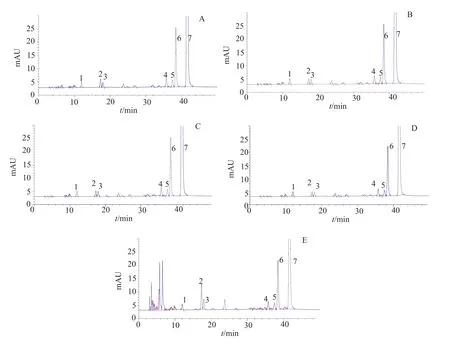
图3 专属性试验色谱图Fig.3 HPLC Chromatograms for specificity test
3.3 检测限(LOD)与定量限(LOQ)
本方法的LOD和LOQ按新霉素B峰的信噪比3:1和10:1计,分别为1.1和3.5ng。
3.4 稳定性
取批号为N-9705-113B的供试品溶液,在18h内每隔6h进样1次,最大杂质峰面积的平均值为1.46%,RSD为0.03%(n=3);杂质总量的平均值为7.5%,RSD为0.23%(n=3),表明供试品溶液在18h内稳定。
3.5 耐用性
分别考察了供试品溶液在Cosmosil C18-PAQ、Zorbax Eclipse plus C18、TSKGEL ODS-100S C18、XBridge C184种不同品牌填料的色谱柱上的分离情况,各杂质之间以及与新霉素B和新霉素C之间均能得到有效分离,说明本方法的耐用性较好。
4 样品测定
采用柱后衍生化-HPLC法对4批硫酸新霉素以及国产与进口对照品按”2.1.1”项下色谱条件及”2.2.1”项下样品浓度进行有关物质检查,按归一化法计算,结果表明,国产硫酸新霉素杂质总量(5.1%~12.3%)大于进口对照品的杂质总量(0.7%~3.8%),同时采用HPLC-PAD法按”2.1.2”项下色谱条件及”2.2.2”项下样品浓度进样分析(表2)。
5 讨论
5.1 分离条件优化
本文采用含烷基磺酸盐的离子对HPLC系统,分别考察了己烷磺酸钠、庚烷磺酸钠和辛烷磺酸钠3种离子对试剂对分离的影响。结果显示,采用相同浓度的己烷磺酸钠,则保留较弱,有关物质在10min内全部洗脱,且几乎均未分离;采用辛烷磺酸钠则保留过强,新霉素B在60min后才洗脱。考虑到分析时间的合理性,选择了庚烷磺酸钠作为离子对试剂。
考察了不同浓度庚烷磺酸钠(5、10和20mmol/L)对色谱分离的影响。浓度较低时(5和10mmol/L),新霉素C与其前相邻杂质共洗脱。浓度为20mmol/L时,各峰之间分离较好,最终确定庚烷磺酸钠浓度为20mmol/L。考察了不同浓度硫酸钠(20、60和110mmol/L)对色谱分离的影响。浓度越高,保留时间越短。综合考虑分离度,分析时间合理性,确定硫酸钠浓度为110mmol/L。
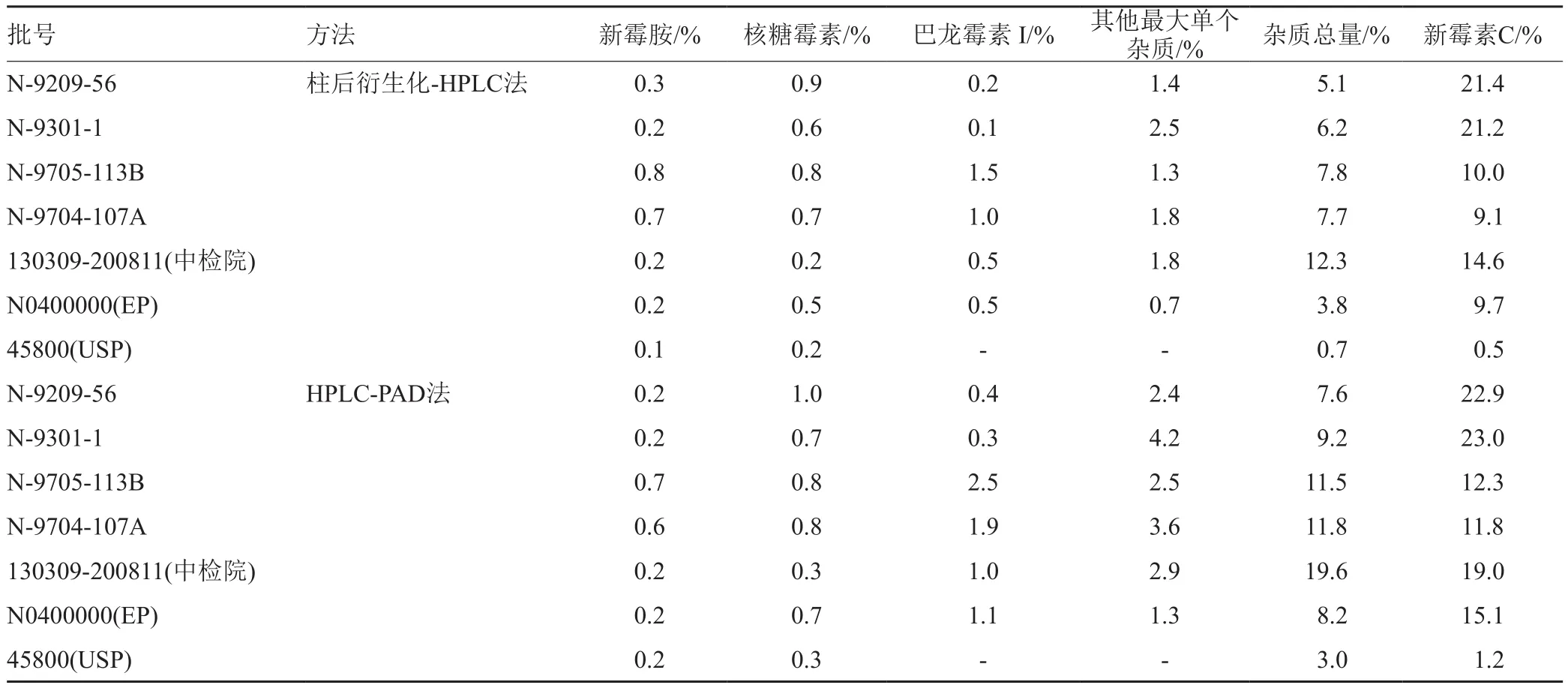
表2 有关物质检测的结果Tab.2 The results of the related substances detection
采用冰醋酸调节缓冲液pH值,考察了pH3.2、3.4、3.8和5.7条件下新霉素B及其有关物质的分离情况。pH在3.2~5.7范围内,pH值对新霉素及其有关物质之间的分离影响极小,pH3.4时,新霉素C与其前相邻杂质具有较高的分离度,新霉素B峰对称性较好。故确定缓冲液pH值为(3.4±0.1)。
考察了流动相中添加乙腈或甲醇对分离的影响。结果显示,采用乙腈可获得更好的分离和更高的柱效,故选择乙腈作为有机改性剂。
考察了色谱柱温对分离的影响,结果显示,降低柱温不仅可改善分离,也可改善新霉素C与其前相邻杂质之间的分离,但柱温降低至20~25℃时,峰形拖尾严重,保留时间过长,故选择柱温为30℃。
5.2 杂质LP-A和LP-B确认
采用制备液相,分别制备HPLC-柱后衍生系统中保留时间约为12和37min的杂质,氮吹富集后,采用文献[5]的HPLC-PAD系统进行确认。结果显示收集的杂质与文献中的杂质LP-A和杂质LP-B峰对应,推断保留时间约为12min的杂质为LP-A,37min的杂质为LP-B。
5.3 HPLC-PAD法与柱后衍生化-HPLC法比较
5.3.1 检测方法的比较
邻苯二甲醛(OPA)与氨基糖苷类抗生素的衍生化反应位点为伯氨基,如果目标物中不含伯氨基基团,则无法进行衍生化,从而不会被紫外活荧光检测器检测到[14]。HPLC-PAD法采用脉冲安培检测器(四电位波形),电活性分子的羟基在工作电极表面发生氧化还原反应,从而产生电极信号而被检测到。
实验结果显示,两批样品(批号:N-9209-56、N-9705-113B)分别在柱后衍生化-HPLC与HPLC-PAD系统中检出的杂质个数相同,即HPLC-PAD系统中检测到的杂质在柱后衍生化-HPLC系统中均可检测到,但两种方法的检测结果存在差异,这与两种检测方法的检测原理不同有关(图4和表2)。
5.3.2 灵敏度比较
HPLC-PAD法的检测限按新霉素B峰的信噪比3:1计,约为1.0ng,与柱后衍生化-HPLC法检测限(1.1ng)相近。柱后衍生化-HPLC法与HPLC-PAD法均具有较高的灵敏度。
5.3.3 实验条件比较
HPLC-PAD法对实验人员的操作技能要求较高;PAD的电极易钝化,需要经常清洗,且清洗电极之后分析系统需要较长的平衡时间[15];实验用到的水和试剂均不得含有电化学活性杂质。而柱后衍生化-HPLC法在仪器及试剂等方面要求较低,具有更好的普适性。
6 结论
本文建立的柱后衍生化-HPLC-荧光检测法操作简便、分离能力强,准确度和精密度均较好,适用于硫酸新霉素有关物质的检测。
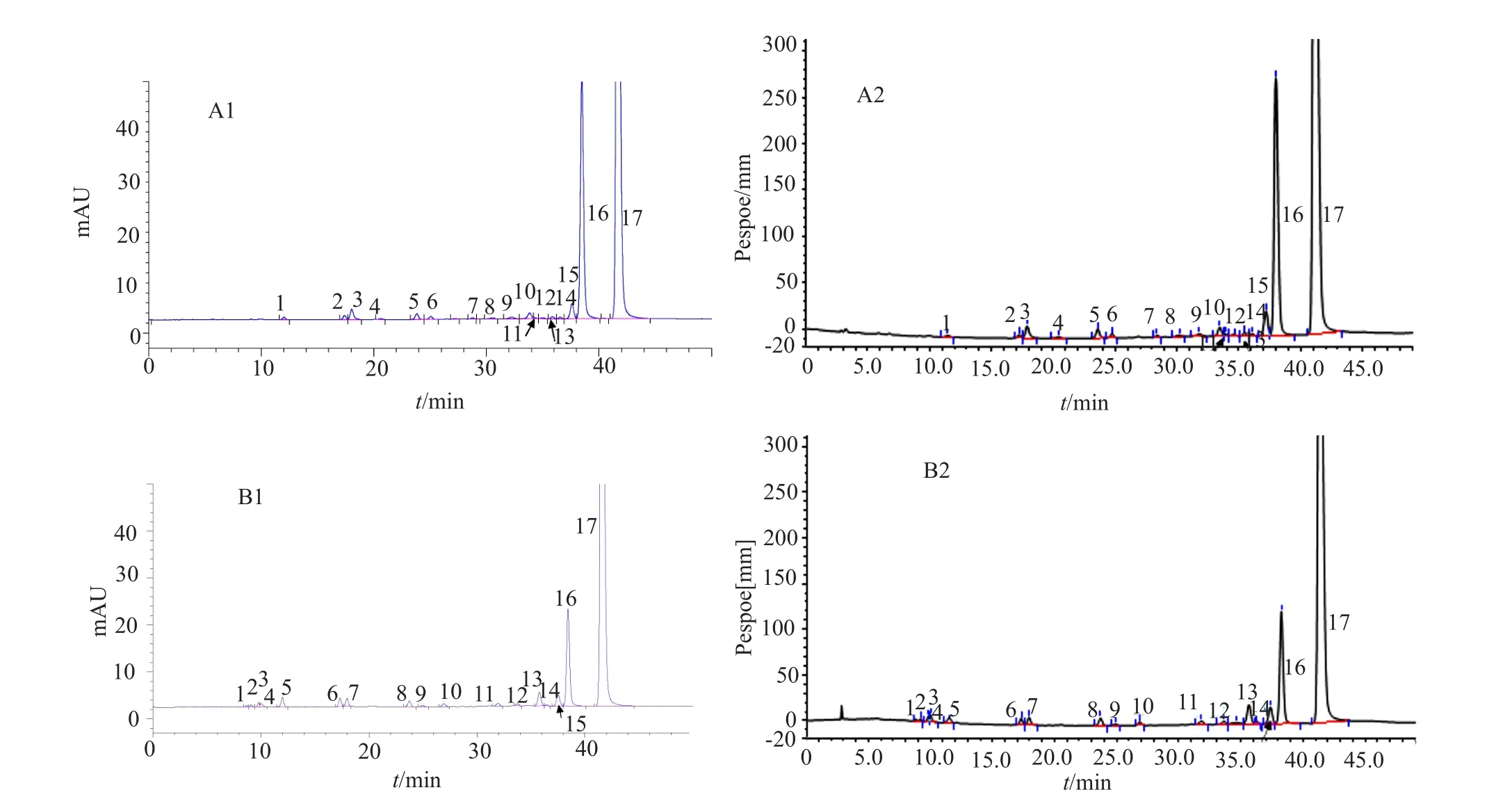
图4 两种分析系统分析硫酸新霉素的图谱Fig.4 The results of analysis of sulfate neomycin with two analytical systems
