抗真菌药物泊沙康唑关键中间体的工艺开发
2019-06-01陆一菱马晴徐敏
陆一菱 马晴 徐敏
(成都市第三人民医院,成都 610031)
泊沙康唑(posaconazole)是伊曲康唑的衍生物,属于广谱三唑类抗真菌药物,由默沙东公司研发、生产;其口服混悬剂于2005、2006年分别被欧洲药品评估局(EMEA)和美国食品药品监督管理局(FDA)批准用于治疗真菌感染,并于2013年获的中国国家食品药品监督管理局的进口注册批准,主要用于预防侵袭性曲霉菌和念珠菌感染包括接受造血干细胞移植(HSCT)后发生移植物抗宿主病(GVHD)的患者或化疗导致长时间中性粒细胞较少症的血液系统恶性肿瘤患者、治疗口咽念珠菌病包括伊曲康唑和/或氟康唑难治疗口咽念珠菌病[1]。随后,FDA又陆续批准了泊沙康唑缓释片、静脉注射液的上市。
目前,作为侵袭性真菌感染患者的一线预防用药,泊沙康唑已经陆续在全球70多个国家上市,为深部真菌感染的预防与治疗提供了一种新的选择。
泊沙康唑因其广谱、高效、低毒的药效特点而被广泛应用于临床,具有广阔的临床应用前景,因此,泊沙康唑自上市以来,受到国内外各大药企、科研院所的青睐,纷纷对泊沙康唑项目进行开发研究,主要包括新工艺、新剂型的开发以及化合物结构修饰等。

图1 泊沙康唑的结构Fig.1 The structure of posaconazole
本文结合泊沙康唑的化学结构特点(图1)进行逆合成分析,并对其关键中间体进行工艺开发,实现了该中间体的工艺开发。该工艺原料易得、反应条件温和、操作简便,有较好的工业化实施基础。
1 泊沙康唑逆合成分析
泊沙康唑属于第二代三氮唑类抗真菌药物,其结构延续了伊曲康唑的三氮唑结构。根据逆合成分析思路并结合其结构特点分析认为,泊沙康唑可以由中间体M1和M2通过SN2取代反应制备;而中间体M2结构中的1,2,4-三唑-3-酮结构可以由中间体M3和M4通过胺解/缩合关环步骤制备。
根据泊沙康唑的逆合成分析(图2)可以看出,其结构中含有4个手性中心,其完全来源于中间体M1和M2,而中间体M2的光学纯度很大程度上取决于中间体M4的光学纯度,即中间体M1和M4的光学纯度决定着泊沙康唑的光学纯度。因此,在泊沙康唑的合成工艺研究时需重点关注中间体M1、M2以及M4的质量,并在泊沙康唑的合成过程中对其手性异构体进行严格控制。
2 关键中间体M4的合成分析
文献检索分析,专利CN106986787A[2]、CN103936564B[3]分别介绍了以手性催化剂、手性还原剂进行还原的方式构建手性中心,手性催化剂/还原剂价格昂贵,工业生产成本较高;而Terashima等[4]报道的路线(图3)以(S)-乳酸乙酯为原料,经胺解、羟基保护、红铝还原、缩合成腙、格氏反应等步骤实现中间体M4的制备;该工艺过程中涉及还原剂红铝的使用,其活性较高,需要低温反应;同时,红铝活性较高可能导致过度还原杂质的产生,反应控制比较繁琐。
专利US5625064[5]、WO9633163[6]报道的路线(图4)是以(S)-乳酸甲酯为原料,经胺解、羟基保护、格氏反应、羰基还原、取代、拆分等步骤实现中间体M4的合成;该工艺过程中涉及硼氢化锂/溴化锌还原,其原位生成硼氢化锌进行还原,硼氢化锂活性较高,易制爆而受管制,不适合工业化实施。同时,文献报道在羰基还原后,与对氯苯磺酰氯反应成磺酸酯,其进一步与水合肼发生SN2取代反应,受磺酸酯活性影响,该步骤收率较低。

图2 泊沙康唑的逆合成分析图Fig.2 The retrosynthetic analysis of posaconazole
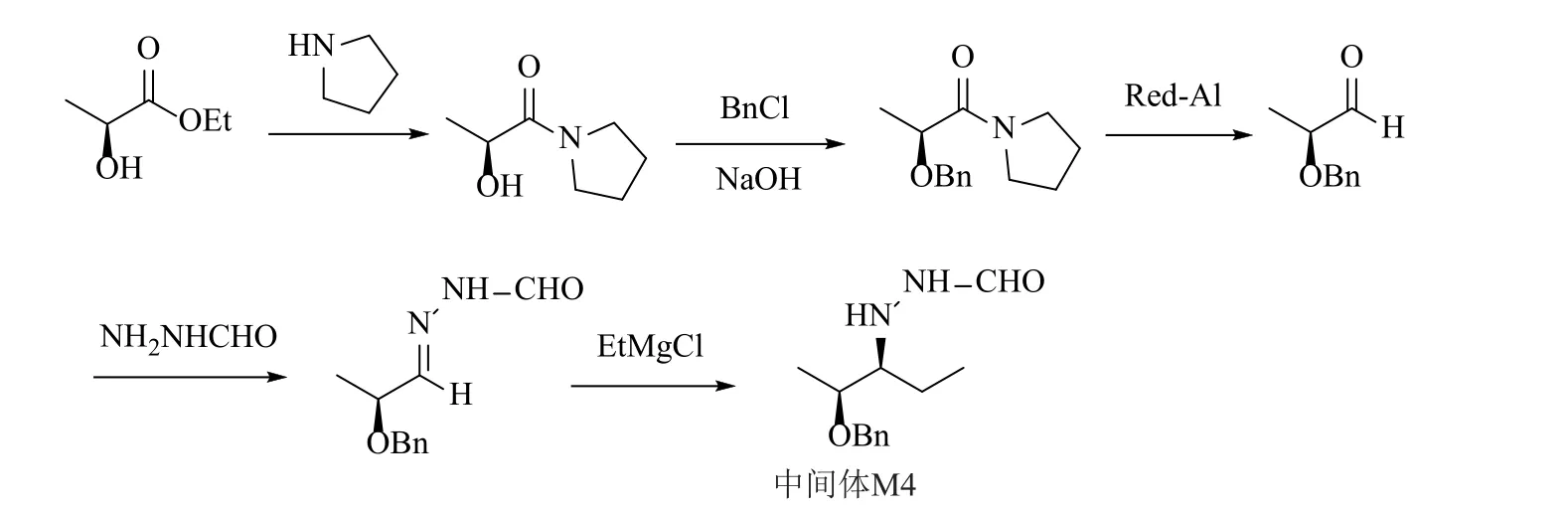
图3 报道的中间体M4的合成路线[4]Fig.3 The synthetic route of intermediate M4 reported[4]
基于文献报道工艺存在的弊端,本研究对中间体M4的合成思路进行了综合分析,以US5625064报道的合成路线为基础进行改进,以硼氢化钠/溴化锌体系替换不能工业化实施的硼氢化锂/溴化锌体系并优化反应条件,取得了很好的反应效果;同时,鉴于对氯苯磺酸酯的活性较低,从而导致后续反应收率较低;本研究以甲磺酰氯代替对氯苯磺酰氯制备甲磺酸酯进行取代反应,其活性增强的同时,其体现出很好的反应活性和立体选择性,取得了很好的反应效果(图5)。
新开发的合成工艺避免了危化品硼氢化锂的应用,反应条件温和,生产过程中易于控制;同时,甲磺酸酯的引入增强了中间体的反应活性,提高了收率;另外,工艺中多步中间体无需纯化,直接用于后续反应,简化了操作步骤;工艺过程经拆分、结晶等纯化步骤,使得中间体M4和M2的光学纯度≥99%。
3 实验部分
3.1 中间体B的制备

图4 专利US5625064报道的中间体M4的合成路线Fig.4 The synthetic route of intermediate M4 reported by US5625064
将10g硼氢化钠悬浮于250mL THF中,冷却至0~10℃,分批加入溴化锌。加完后,维持-5~5℃搅拌1h。将溶有45g中间体A(0.23mol)的100mL THF溶液(中间体A的制备可参考US5625064以S-乳酸甲酯为原料制备)缓慢滴入上述反应液中,滴完后维持-5~5℃反应。TLC监控反应完全。依次向反应液中滴加70mL丙酮、90mL水终止反应;继续向反应体系中滴加50mL 2mol/L盐酸溶液。浓缩蒸出THF,加入EA萃取3次,合并有机层,有机层依次用水洗、饱和食盐水洗,无水硫酸钠干燥,过滤浓缩得到中间体B粗品(黄色油状物)41g,收率:90%。无须纯化直接用于下一步。
3.2 中间体C的制备
依次向上述中间体B粗品中加入200mL二氯甲烷、40g DMAP,搅拌溶清后,冷却至0~5℃,将35g甲磺酰氯(0.30mol)缓慢滴入反应液中,滴完后维持0~5℃反应。TLC监控反应完全。向反应体系中加入200mL水,分出有机层,水层用100mL二氯甲烷萃取两次,合并有机层,有机层依次用10% NaOH溶液、稀盐酸、饱和食盐洗涤,浓缩得到中间体C粗品,黄色油状物47g,收率:82%,无须纯化直接用于下一步反应。
3.3 中间体D的制备

图5 新设计的中间体M4及中间体M2合成工艺路线Fig.5 The newly designed synthetic route of intermediate M4 and M2
向上述中间体C粗品中加入100mL乙醇溶解,搅拌条件下将50mL水合肼加入反应液中,缓慢升温至75℃搅拌反应过夜。TLC监控反应完全。浓缩蒸出乙醇,加入60mL水,用20mL×2乙酸乙酯萃取,合并有机层,有机层用饱和食盐水洗、干燥、过滤。向滤液中加入36g柠檬酸(0.19mol),搅拌析晶1h,过滤得中间体D消旋体的柠檬酸盐,然后用碳酸钾中和酸,EA萃取得到游离碱精制品22g[1H NMR确定(2S,3S):(2S, 3R)非对映异构体比例为88:12]。
将上述中间体D消旋体溶于100mL 叔丁基甲基醚中,将溶有38gL-二苯甲酰基酒石酸(L-DBTA)的叔丁基甲基醚溶液(300mL叔丁基甲基醚)加入上述反应液中,反应液中缓慢析出固体,搅拌析晶2h,过滤,MTBE洗涤,重结晶得到中间体D 45g[1H NMR确定(2S, 3S):(2S, 3R)非对映异构体比例为99:1],3步总收率为34%。
3.4 中间体M4的制备(图6)
将40g中间体D加入400mL甲酸乙酯中,搅拌加热至回流反应,固体溶解。TLC监控反应完全。降温后,向反应液中加入200mL甲基叔丁基醚和碳酸钠饱和溶液,分层,萃取有机相,碳酸氢钠溶液洗涤,饱和食盐水洗,干燥浓缩得到中间体M4 14g,收率为84%;纯度:96%,光学纯度为99.4%(图6)。异构体方法:按照高效液相色谱法(中国药典2015年版四部通则0512)测定,用CHIRALCEL OD(4.6mm×250mm, 5μm)手性色谱柱,以正己烷-异丙醇(85:15)为流动相;检测波长为215nm,柱温25℃,流速1.0mL/min。1H NMR(d6-DMSO,400MHz):δ=7.98(s, 1H), 7.42~7.29(m, 5H), 4.69(d,J=8.0Hz,2H), 3.50~3.47(m, 1H), 2.54~2.53(m,1H)1.52~1.36(m, 2H), 1.27(d,J=8.0Hz 3H), 0.95(t,J=8.0Hz, 3H)ppm;MS-ESI(m/z): 237.5[M+H]+。
3.5 中间体M2的制备(图7)

图6 中间体M4的光学纯度检测图谱Fig.6 The detected optical purity of intermediate M4
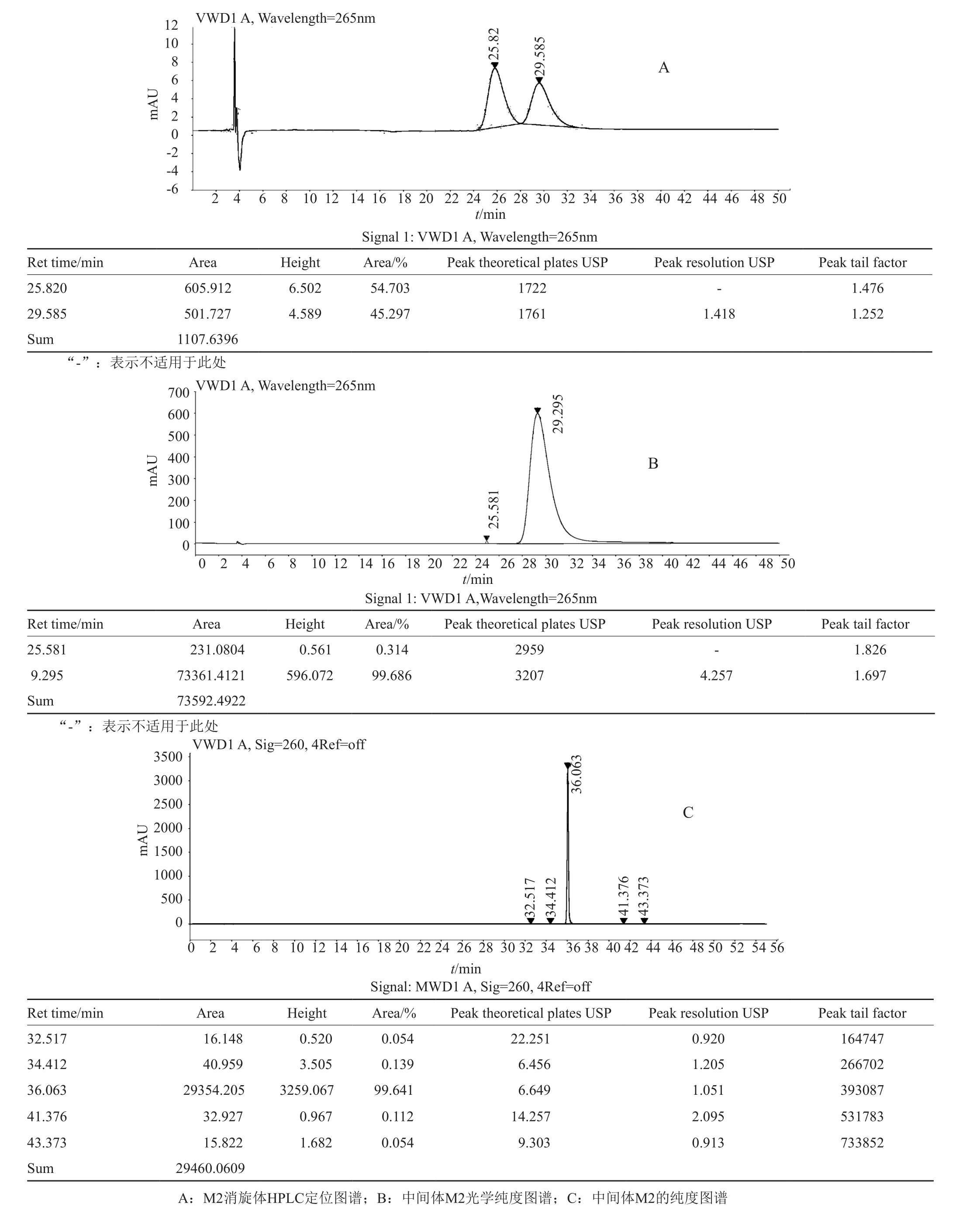
图7 中间体M2的纯度和光学纯度检测图谱Fig.7 The detected purity and optical purity of intermediate M2
依次将1 0 g中间体M 4、1 3 g中间体M3(33.4mmol),100mL二氧六环加至反应釜中,搅拌升温至80℃,缓慢滴加5g三乙胺,滴完后维持该温度搅拌反应。TLC监控反应完全。将反应液降温至室温,向反应液中加入150mL水,搅拌1h,过滤,固体用水洗,干燥后将固体溶于三氯甲烷中,活性炭脱色,过滤,滤液减压浓缩得到中间体M2粗品。
向中间体M2粗品中加入200mL甲苯,升温至100℃搅拌2h,然后冷却至10℃析晶2h,过滤固体,干燥得中间体M2 13g,收率为74%;纯度:99.6%,光学纯度为99.7%(图7)。异构体方法:按照高效液相色谱法(中国药典2015年版四部通则0512)测定,用CHIRALCEL OJ(4.6mm×250mm, 5μm)手性色谱柱,以正己烷-异丙醇(70:30)为流动相;检测波长为265nm,柱温25℃,流速1.0mL/min;有关物质检测方法:按照高效液相色谱法(中国药典2015年版四部通则0512)测定,用氰基柱[如Kromasil 60-5-CN(4.6mm×250mm, 5μm),以10mmol/L磷酸氢二钾溶液(用磷酸调节pH值至5.0)为流动相A;以乙腈为流动相B,按照A:B为(90:10-25:75-90:10)的比例进行梯度洗脱,检测波长为260nm,柱温为40℃,流速为1.0mL/min]。HRMS-ESI(m/z): 514.2797 [M+H]+;1H NMR(d6-DMSO, 400MHz):δ=8.88(s, 1H),8.33(s, 1H), 7.50~6.69(m, 13H), 4.56~3.73(m, 4H),3.35~3.11(m, 8H), 1.79~1.74(m, 2H), 1.24(d,J=8.0Hz 3H), 0.80(t,J=8.0Hz, 3H)ppm.
4 讨论
本文基于文献报道的工艺特点并结合泊沙康唑关键中间体M4和M2的结构特性,对其合成工艺进行科学、系统地分析,评估其潜在的异构体残留等风险;根据分析结果对泊沙康唑关键中间体M4及M2的合成路线进行设计、优化,避免了危化品应用的同时,提高了反应中间体的反应活性,实现了对生产工艺、产品质量的有效控制。同时,该工艺中多步反应中间体无需纯化,直接用于后续反应,简化了操作步骤,具有反应原料易得、反应条件温和、操作简便等特点,具有较好的工业化实施基础。
