银芩胶囊HPLC指纹图谱研究
2019-05-29
1.昆明医科大学药学院暨云南省天然药物药理重点实验室,云南 昆明 650500;2.昆药集团股份有限公司,云南 昆明 650100
银芩胶囊是在傣族“雅哇黄”方基础上开发的现代制剂[1],其主要由黄芩、金银花、鱼腥草、三七叶合理配伍而成,其内容物为黄色至黄褐色颗粒和粉末,气微、味苦。本品具有清热解毒之功效,用于治疗外感风热所致的发热、咳嗽、咽痛及呼吸道感染、扁桃体感染和咽炎[2]。黄芩中黄酮及其苷类是其主要的药效成分[3],金银花化学成分主要包括有机酸、黄酮类、三萜皂苷类、环烯醚萜类、挥发油及微量元素等,其中有机酸以绿原酸类化合物为主[1,4]。三七叶中富含皂苷类成分[5]。鱼腥草中挥发类成分较多,非挥发性成分较少,非挥发性成分主要有槲皮苷、芦丁、金丝桃苷等[6]。中药复方制剂通常由多味药组成,其中所包含的化学成分上百种,这给复方制剂的研究带来一定的困难。中药指纹图谱[7]是一种综合的、可量化的分析手段,是目前符合中药特点、评价中药质量的控制模式之一。指纹图谱能反映中药制剂的整体特征,提高中药复方制剂质量控制的专属性,在鉴别和评价药品内在质量方面的作用远大于选择一个指标成分测定含量的作用[8-9]。因此更为科学和合理,也更符合中药的多途径协调作用的药理特点。
1 实验材料
1.1 仪器与试药 Waters RD.LC 高效液相色谱仪,美国Agilent公司;Milli-Q 超纯水器,Millipone 公司;XP 205分析电子天平,梅特勒-托利多公司;X Bridge C18色谱柱(4.6 mm×250 mm,5 μm)。黄芩苷对照品(批号MUST-18010410,Purity 98.58%,HPLC),中国科学院成都生物研究所;银芩胶囊(批号:161009-01、161009-02、170605-01、170605-02、180401-01、180401-02、180206-01、180206-02、180307-01、180307-02),昆药集团股份有限公司;
2 方法与结果
2.1 供试品溶液的制备 精密称取芩胶囊内容物0.5 g,置于具塞锥形瓶中,加70%甲醇10 mL,超声处理20 min(功率250 W,频率350 Hz),进行提取两次,再倒入10 mL离心管中离心15 min,经0.45 μm微孔滤膜过滤,取续滤液即得。
2.2 对照品溶液的制备 取黄芩苷对照品,加70%甲醇制成每1 mL中含100 μg的溶液,即得。
2.3 色谱条件 色谱柱:X Bridge C18色谱柱(4.6 mm×250 mm,5μ m);流动相:乙腈(A)-0.1%甲酸水(B),以表1中梯度洗脱条件洗脱;柱温25 ℃;检测波长:280 nm;流速1.0 mL/min。
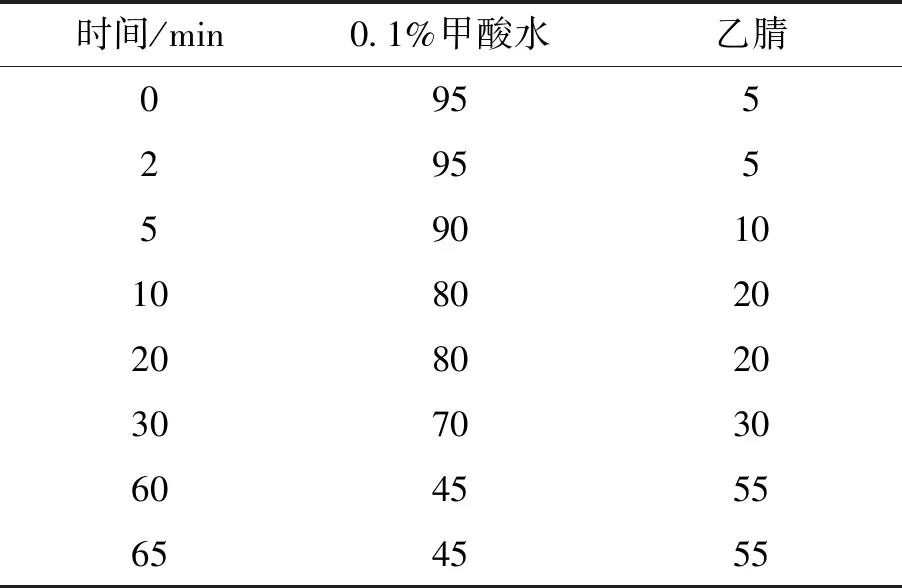
表1 梯度洗脱表
2.4 方法学考察
2.4.1 仪器精密度试验 取同一供试品溶液,按“2.3”项下色谱条件进样6次,测得各共有峰的相对保留时间RSD<0.3%,各共有峰相对峰面积<0.13%合指纹图谱技术标准的要求,结果显示方法的精密度好。见表2。
2.4.2 重复性实验 取同一批银芩胶囊样品,按“2.1”项下方法制备6份供试品溶液,按“2.3”项下色谱条件进样测定,测得各共有峰的相对保留时间RSD<2.1%,各共有峰的相对峰面积RSD<0.12%。表明该方法重复性良好,符合指纹图谱的测定要求。见表3。
2.4.3 稳定性试验 取同一批供试品溶液,分别于0、6、12、24、36、48 h进行指纹图谱测定,测定各共有峰的相对保留时间RSD<0.32%,各共有峰的相对峰面积RSD<0.89%,表明供试品溶液在48 h内稳定性良好。见表4。
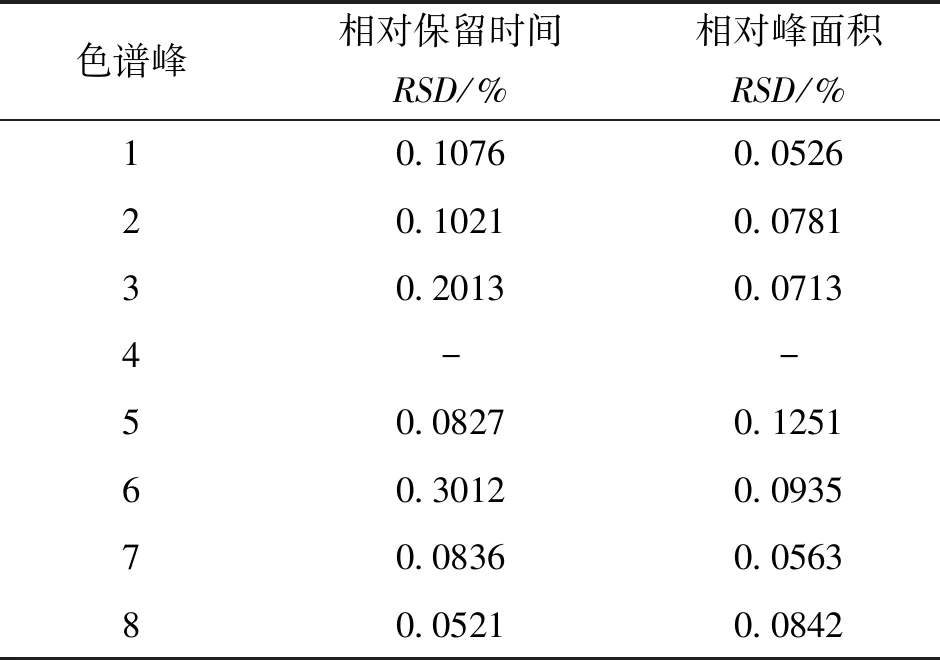
表2 银芩胶囊共有峰精密度
注:4号峰(黄芩苷)为参照峰。
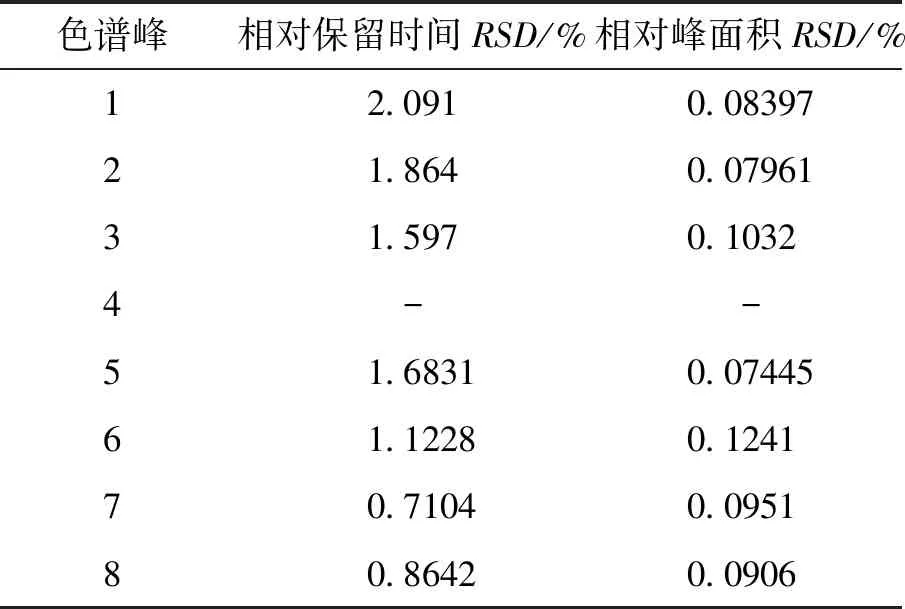
表3 银芩胶囊共有峰重复性
注:4号峰(黄芩苷)为参照峰
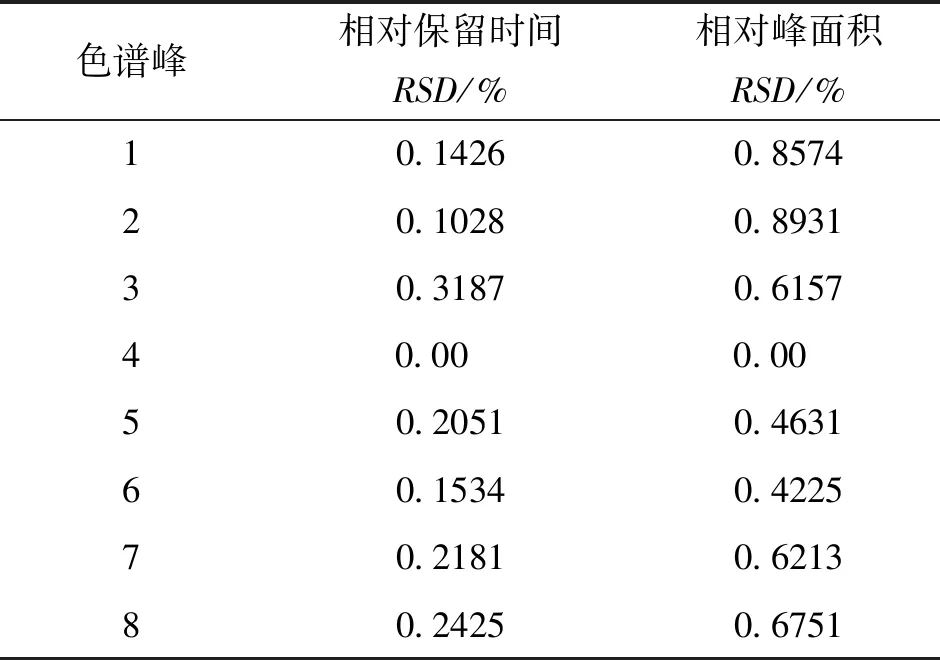
表4 银芩胶囊共有峰稳定性
注:4号峰(黄芩苷)为参照峰。
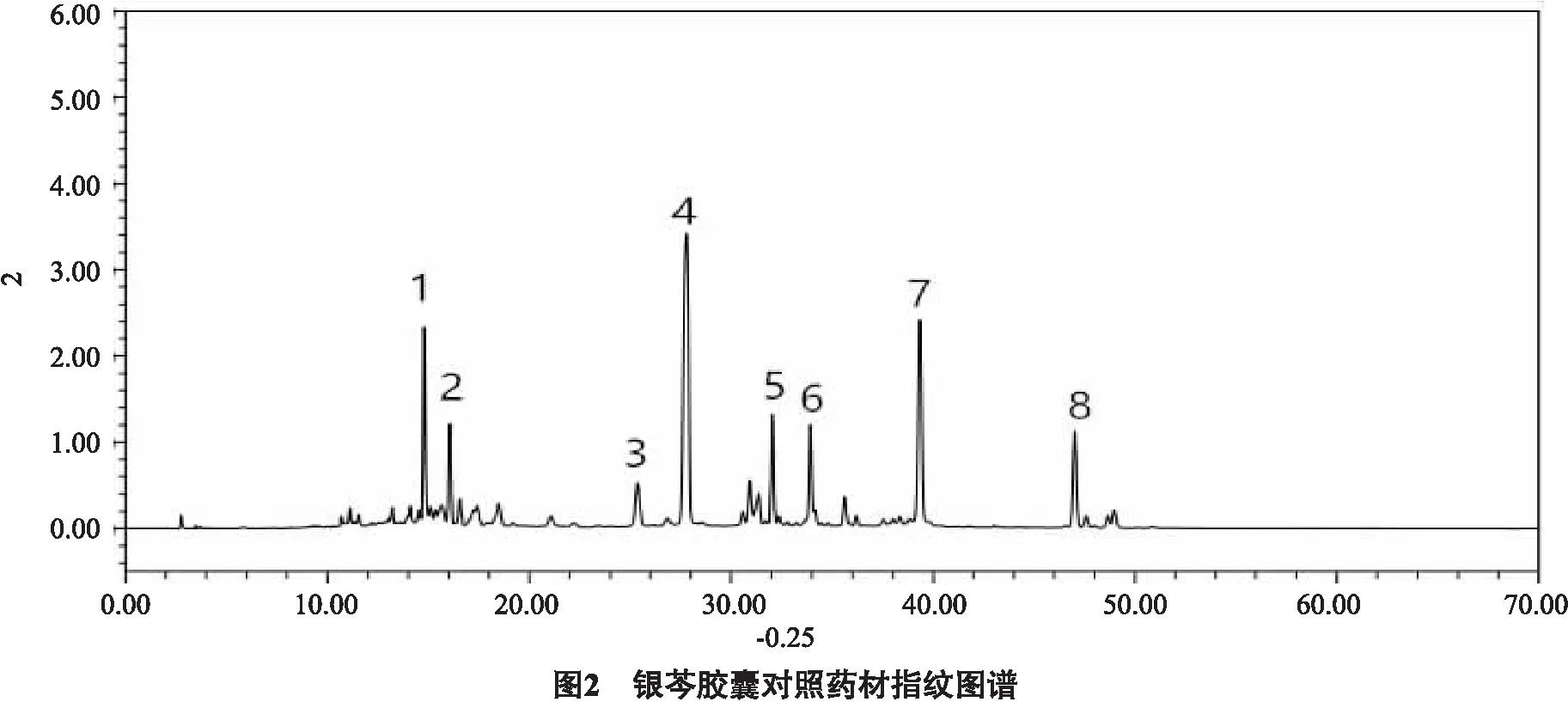
2.5 指纹图谱的建立及共有峰的标记 分别制备10批昆药集团生产的银芩胶囊供试品溶液,按“2.3”项下色谱条件进行测定,色谱图如图1所示;各批银芩胶囊指纹图谱概貌一致,相似度值均大于0.98,总体相似度较高,见表5;选取8个共有峰作为特征指纹峰,建立银芩胶囊对照指纹图谱,如图2所示。
分析十批银芩胶囊HPLC色谱图,峰4为黄芩苷,峰6为汉黄芩苷,峰7为黄芩素,峰8为汉黄芩素,其中峰4在色谱图中分离效果较好,含量高,且为银芩胶囊中药的质量控制指标成分,故确定峰4黄芩苷为参照峰。
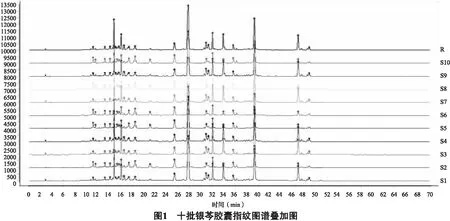
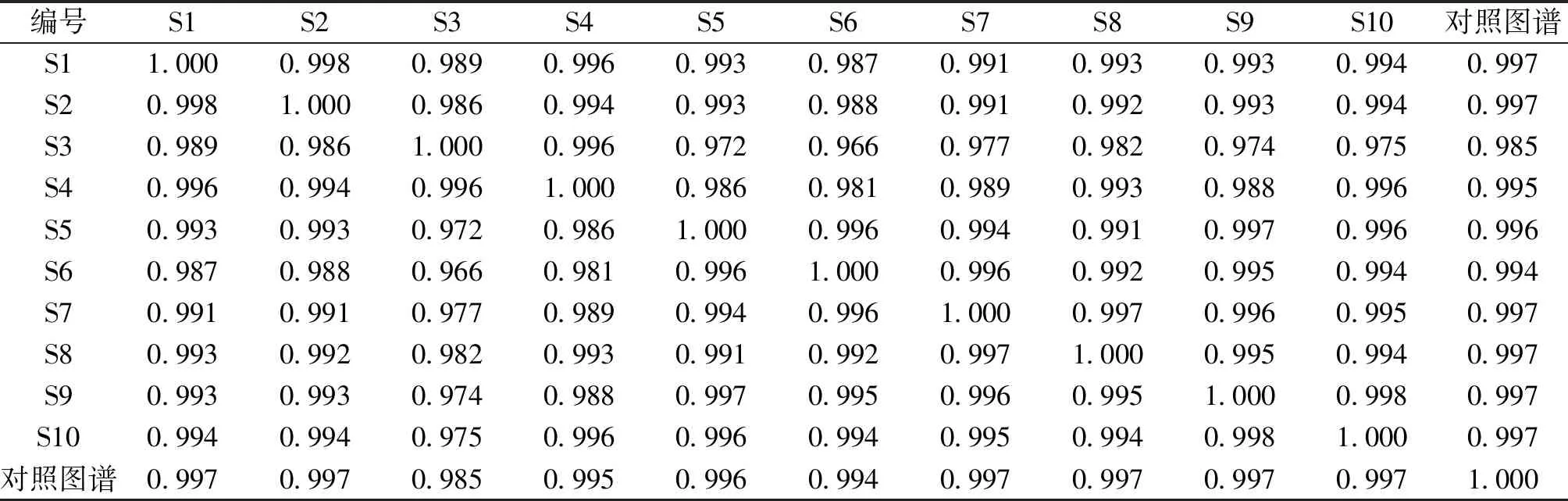
表5 十批银芩胶囊指纹图谱相似度评价
3 讨论
3.1 固定相的选择 本试验比较了不同厂家不同型号的3种色谱柱:①Ulatimate AQ C18(4.6 mm×250 mm,5 μm);②LUNA C18(4.6 mm×250 mm,5 μm);③X Bridge C-18(4.6 mm×250 mm,5 μm);结果表明色谱柱①的分离效果较差,黄芩苷未能和小峰分离,且峰形不好。色谱柱③的黄芩苷峰也未能达到分离;色谱柱②的分离效果均良好,故选择X Bridge C18色谱柱。
3.2 流动相体系的选择 取同一供试品溶液,考察不同流动相体系对色谱峰分离效果的影响,采用甲醇-水流动相体系,因甲醇洗脱能力较弱,色谱峰峰形不好且出峰不完全。故考察了乙腈-水流动相体系,乙腈-0.1%甲酸水流动相体系、乙腈-0.1%乙酸水流动相体系。试验过程中,采用梯度洗脱分离银芩胶囊中的有效成分,结果表明,当流动相为乙腈-0.1%甲酸水时,色谱峰出峰完全且峰形较好。故选择乙腈-0.1%甲酸水溶液。
3.3 梯度的选择 取同一供试品溶液,分别考察了三个梯度洗脱条件对色谱峰分离效果的影响,发现只有在表1梯度洗脱条件下,黄芩苷的峰能与其他小峰分开。因此选择表1中的梯度条件进行洗脱。
3.4 检测波长的选择 取同一供试品溶液,以表1中梯度洗脱条件洗脱,进行检测波长的选择考察,取同一供试品溶液,测定某一特定成分含量的检测波长通常不一定适合指纹图谱的需要,为了获得多层次的信息,需要选择数个不同的检测波长,同时银芩胶囊为复方制剂,其所含化学成分较多。因此,在试验中应使用PDA检测器全波长检测获得3D指纹图谱[10]。结果表明280 nm处可以得到较多的化学信息,故选择280 nm作为检测波长。
3.5 柱温的选择 取同一供试品溶液,考察不同色谱柱温度(20 ℃、25 ℃、30 ℃)的色谱峰分离情况,测得三种不同温度下色谱分离效果相似,因为25 ℃接近室温,最终确定25 ℃为测定用柱温。
4 结论
本试验通过供试品前处理方法的研究,色谱条件的研究,最终确定银芩胶囊指纹图谱的分析方法,因目前银芩胶囊仅在昆药集团生产,因此产品批数较单一,后续还需大量样品的质量跟踪,数据积累,初步暂定银芩胶囊的指纹图谱相似度在0.90以上为合格。本研究尝试了多种不同梯度的洗脱条件,最终确定了银芩胶囊的指纹图谱分析的色谱条件,考察确定了8个共有峰的对照指纹图谱,方法快速、简便、重复性好,经方法学验证满足其相关要求。本研究建立的一种银芩胶囊的指纹图谱分析方法适用性强,能够全面反映产品的质量状况。
