32例智力低下发育迟缓患儿基因组拷贝数变异的分析
2019-05-24党颖慧宋婷婷万陕宁郑芸芸陈必良张建芳
党颖慧 宋婷婷 万陕宁 郑芸芸 黎 昱 陈必良 张建芳
儿童智力低下(mental retardation,MR)和发育迟缓(developmental delay,DD)是一类严重危害儿童健康的疾病,发生率约为 2.3%[1]。在我国导致智力低下的先天性因素中,遗传病约占 40.5%,而染色体因素占遗传因素的 37.0%[2]。染色体微阵列分析技术(CMA)是一种全新的分子核型分析技术,具有敏感度高、分辨率高、自动化程度高等优点,在遗传性疾病和正常人群中能检测出各种基因拷贝数变化,应用CMA对CNVs进行深入研究是国内外研究该病遗传机制的发展趋势,但国内应用经验仍有限[3]。本研究应用CMA对32例不明原因的MR/DD患儿进行全基因组扫描与分析,探讨CMA对不明原因MR/DD患儿可能的分子病因诊断的作用。
对象与方法
1.筛选标准:选择2015年1月~2017年11月到笔者医院儿科及遗传咨询门诊就诊的患儿32例为本次调查对象,选择标准包括:①临床诊断为智力低下/发育迟缓患儿;②无明显围生期异常病史,无明确出生后缺氧、中毒、头颅外伤及中枢神经系统感染等病史;③无甲状腺功能异常,无颅内肿瘤;④常规染色体核型正常;⑤排除已知的遗传性综合症和遗传性代谢病导致的智力低下。其中男性患儿14例,女性患儿18例,患儿年龄2个月~10岁,主要表现为智力低下、生长发育迟缓、特殊面容等。根据不同的年龄段分别采用《Gesell发育量表》和《中国韦氏智力量表》测定儿童的发育商(DQ)或智商(IQ),同时运用《婴儿-初中生社会适应性能力量表》进行社会适应性能力评价。IQ或DQ<70,伴社会适应性能力低下者诊断为MR,5岁称为MR,<5岁称为DD;以下统称为MR/DD。告知所有患儿监护人CMA的检测范围及局限性,签署书面知情同意书。本研究获得笔者医院医学伦理学委员会批准。
2.检测方法:皮肤表面常规消毒后抽取静脉血2ml,使用 DNA 提取试剂盒(QIAamp DNA Blood Mini Kit)按说明书操作方法提取基因组DNA。应用Affymetrix公司的Cytogenetic CytoScan 750K Array全基因芯片扫描技术检测全基因组CNVs。Affymetrix CytoScan 750K Array芯片包含有20万SNP标记和55万CNV标记,以平均大约1marker/4kb的密度分布于人类的整个基因组,用于检测全基因组染色体。按试剂盒说明书操作:将DNA溶解于AE buffer 溶液并将DNA 浓度调整至50ng/μl。对DNA样品进行酶切、连接、PCR扩增。将扩增产物用磁珠法进行纯化,纯化后的PCR产物进行片段化处理,片段在25~125bp。片段化后的产物在末端脱氧核糖转移酶(TdT)的作用下进行标记反应。标记完成后和杂交试剂混合,95℃ 10min,49℃孵育2min后取200μl样本载入到置密的芯片中,密闭50℃,60r/min杂交16~18h。然后将芯片放入洗染工作站内完成洗涤、SAPE染色等步骤。洗染结束后将芯片放入扫描仪进行扫描。用Affymetrix GeneChip Scaner生成原始cel文件通过AGCC软件芯片图像显示分析,利用CHAS软件对结果进行分析。
3.CMA检测结果判定:对于检测出的CNVs通过与公共数据库UCSC、ISCA、DGV、DECIPHER、OMIM等数据库进行比对分析判断其性质,结果分为:①致病性CNVs;②意义不明确CNVs;③意义不明确,可能致病CNVs;④意义不明确,可能良性CNVs;⑤良性CNVs[4]。
结 果
1.32例MR/DD患儿的染色体拷贝数异常情况:在32例不明原因的MR/DD患儿中,共检出16例存在CNVs,临床表现详见表1。其中致病性CNVs 8例(25%,8/32),意义不明确CNVs 6例(19%,6/32),意义不明确、可能良性CNVs 2例(6.3%,2/32)。
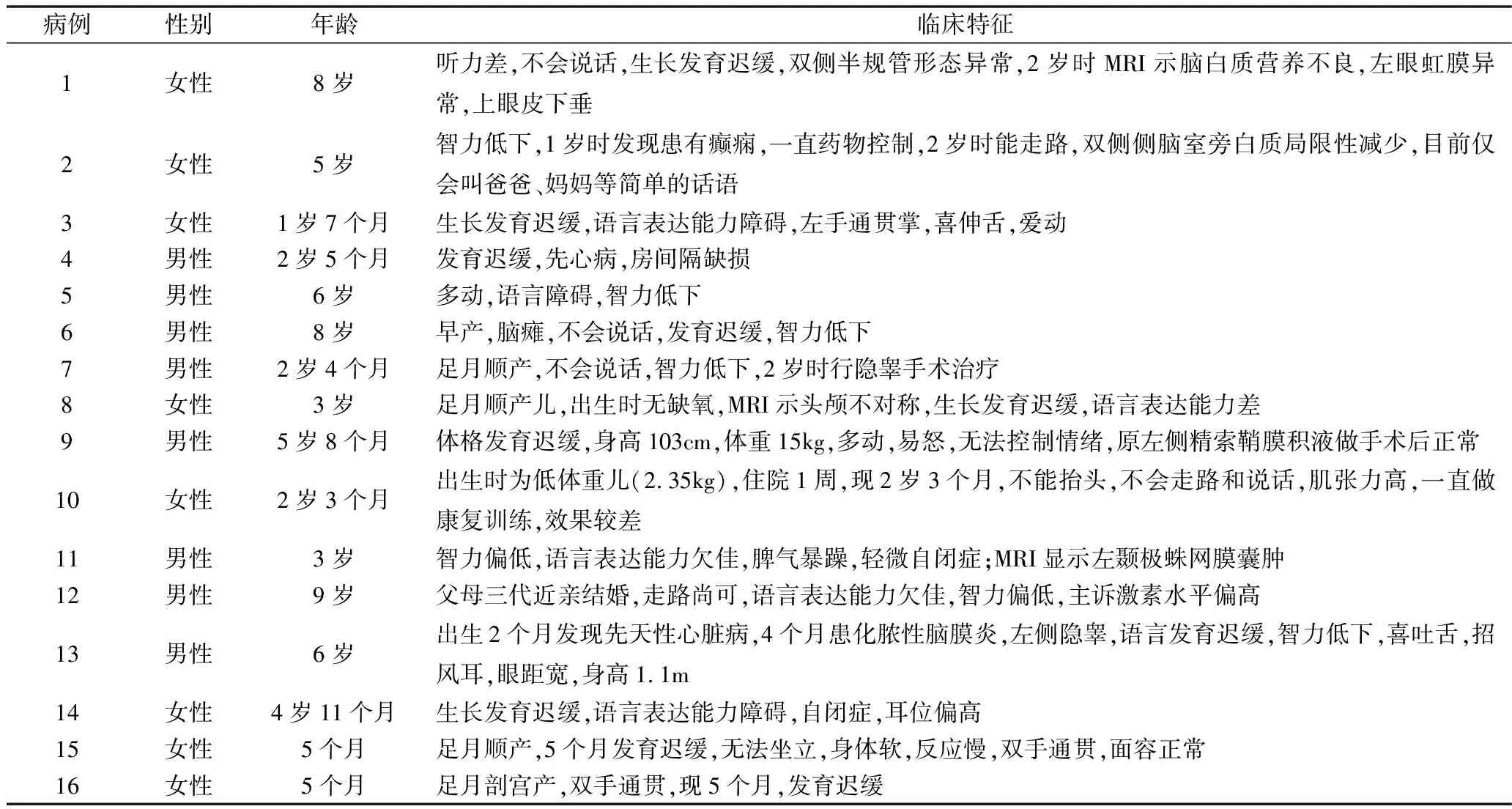
表1 16例MR/DD患儿临床表现
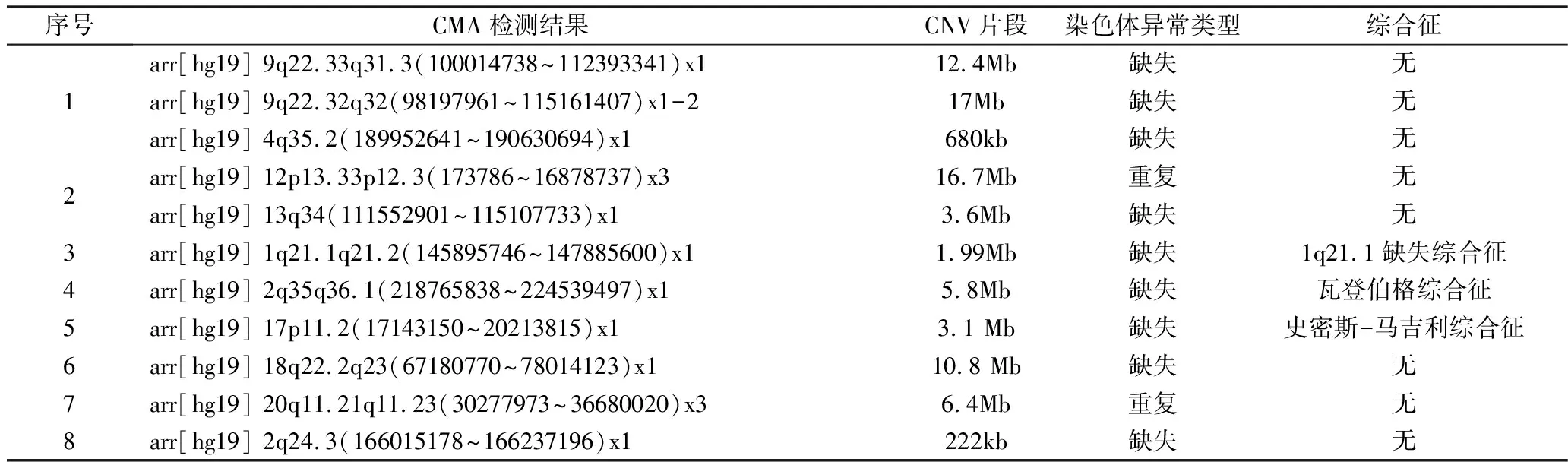
表2 CMA检出的8例致病性CNVs结果对比
2.16例患儿CNVs结果分析:16例患儿中存在CNVs,8例为致病性CNVs,阳性率25%,其中存在缺失6例,重复1例,重复合并缺失1例,所检出的CNVs大小为(0.22~17.00)Mb;3例确诊为已知综合征患儿,分别是1q21.1缺失综合征(1q21.1 deletion syndrome),瓦登伯格综合征(Waardenburg syndrome),史密斯-马吉利综合征(Smith-Magenis syndrome),详见表2。意义不明确CNVs 6例(19%,6/32),其中缺失2例,重复1例,杂合性缺失3例,所检出的CNVs大小为(0.42~19.50)Mb,>10Mb的片段有6个,详见表3。另外意义不明确,可能良性有2例。

表3 CMA检出的意义不明确CNVs结果对比
讨 论
智力障碍是一类遍及全世界的严重危害儿童身心健康的一类疾患,初步估计目前约15%~20%的智力障碍是由于亚显微的染色体拷贝数变异造成的。国外研究已证实在不明原因MR/DD患者中10%~20%存在基因组不平衡,并认为这种基因组失衡是MR/DD甚至神经精神疾患的致病原因之一[5]。G显带染色体核型分析作为首选技术已广泛用于染色体异常的检测,但是其分辨率有限,而CMA能够在全基因组水平进行扫描,尤其是对于检测染色体组微小缺失及重复等不平衡性重排具有突出优势,2010年细胞分子遗传学芯片国际标准(International Standard CytogenomicArray,ISCA) 协会在研究了21698例具有异常临床表征,包括智力低下、发育迟缓、多种体征畸形以及自闭症的先证者的基础上,发现CMA对致病性CNV的检出率为12.2%,比传统G显带核型分析技术的检出率提高了10%。美国医学遗传学会建议,将CMA作为生长发育迟缓、智力低下和孤独症谱系疾病的一线诊断技术,而不再使用传统的显带核型分析技术[6]。
本研究应用CMA检测不明原因MR/DD患儿32例,检测出16例患儿存在CNVs,其中8例发现致病性CNVs,阳性率25%(8/32)。利用CMA在儿科遗传病的临床应用专家共识中针对智力落后和(或)发育迟缓患者进行CNVs检测,阳性率约为19.2%,二者数据结果基本一致[7]。
在32例患儿中,病例1至病例8检测结果为致病性CNVs。病例1检出9号染色体9q22.33q31.3区域有约12.4Mb的缺失,9q22.32q32区域约17Mb的嵌合缺失,该缺失区域包含多个OMIM基因,数据库显示可能与特殊面容、牙齿发育不全,小脑畸形,听力受损相关,此外4号染色体4q35.2区域约680kb的缺失,但没有OMIM基因,有病例报道该区域的缺失与克罗恩病相关,染色体4q缺失综合征是一种罕见的疾病(1∶100000的发生率),具有不同的临床表型,取决于涉及的区域,其包括发育迟缓和异型[8]。4q33区域已被提议为4q缺失综合征的关键区域,而涉及4q34~4q35的更远端缺失与轻度畸形、表型和智力障碍有关[9]。病例2检出12号染色体12p13.33p12.3区域有约16.7Mb的重复,13号染色体13q34区域3.6Mb的缺失。12p13.33p12.3区域含有RAD52、WNK1和HSN2等178个OMIM基因,Decipher数据库显示该区域的重复可能与发育迟缓,智力低下相关。
13q缺失综合征(OMIM:613884)是以生长发育迟缓、智力障碍、先心病和癫痫为特征的染色体病症,不同区域不同位置临床表现不同,长臂末端13q33~34,主要与严重的智力障碍相关,此13q34区域3.6Mb的缺失包含18个OMIM基因,Decipher数据库及相关文献显示该区域缺失与语言障碍、发育迟缓、先心病及智力低下等临床症状有关[9,10]。病例3检出1号染色体1q21.1q21.2区域有约1.99Mb的缺失,该缺失区域包含13个OMIM基因,且与OMIM数据库中的1q21.1缺失综合征(1q21.1 deletion syndrome,OMIM:612474)有重叠区域,有文献提示该区域缺失患者与喂养困难、生长发育迟缓、智力低下、小头畸形等临床表现相关。1q21.1缺失综合征患者表现多样化,大多患者伴有轻到重度的发育迟缓,先天性多发畸形[11]。病例4检出2号染色体2q35q36.1 区域有约5.8Mb的缺失,该区域包含PAX3、DES、EPHA4等52个OMIM基因,ISCA、Decipher、Clinvar等数据库显示该缺失具有致病性,与语言发育迟缓、智力低下、先心病等有关。该缺失区域包含PAX3基因,此基因的缺失或突变会引起瓦登伯格综合征(Waardenburg syndrome,OMIM:193500),它是一种临床罕见的常染色体显性遗传性疾病,主要表现为皮肤、毛发和眼睛的色素异常、感觉神经性耳聋及其他畸形等临床表现。有报道该基因缺失与先心病也有关[12]。
此外2q33.2q37.3区域有约36Mb的嵌合缺失。病例5检出17号染色体17p11.2区域有约3.1Mb的缺失,包含COPS3、NT5M、RAI1等31个OMIM基因,该缺失区域与史密斯-马吉利综合征(Smith-Magenis syndrome,OMIM:182290)区域有重叠,包含的剂量敏感型RAI1基因为该综合征的关键基因,Decipher、ISCA、Clinvar等数据库及文献显示该区域缺失的患者大多数伴有智力低下、睡眠障碍、自我伤害行为、语言表达能力障碍、牙齿异常等临床表现[13,14]。病例6检出18号染色体18q22.2q23区域有约10.8Mb的缺失,该缺失区域包含GALR1、MBP、TSHZ1等24个OMIM基因,Clinvar、Decipher、OMIM等数据库及文献显示该区域缺失的患者可能有发育迟缓、颅面部异常、身材矮小、张力减退、语言表达能力障碍、听力损伤、腭裂等临床表现[15]。病例7检出20号染色体20q11.21q11.23区域有约6.4Mb的重复,该重复区域包含ASXL1、DNMT3B、EPB41L1等68个连续的OMIM基因,Decipher、ISCA、Pubmed等数据库显示该区域重复较为罕见,但有该区域重复致病的病例报道,该区域重复的患者可能有发育迟缓、内眦赘皮、颅面部异常、语言表达能力差等临床表现[16]。病例8检出2号染色体2q24.3区域有约222 kb的缺失,该缺失区域包含SCN3A和SCN2A 2个OMIM基因,这两个基因均为钠离子通道α亚基基因家族成员,SCN2A基因杂合突变或缺失与癫痫性脑病及自闭症相关,有文献报道显示该区域缺失的患者可能有癫痫、自闭症、神经系统异常、精神阻滞、小头畸形等临床表现[17,18]。
本研究32例患儿中,检测出意义不明确CNVs 6例(19%,6/32),意义不明确、可能良性CNVs 2例(6.3%,2/32)。其中病例9~病例14结果为意义不明CNVs,病例15和病例16结果为意义不明确、可能良性CNVs,因为缺失区域包含着丝粒位置且探针较少。病例9检出Y染色体Yq11.223q11.23区域约2.2Mb的缺失,Yq11.23区域约885kb的缺失,该缺失区域与国际公共良性CNVs数据库(DGVs)不重叠,包含DAZI、DNZ2等OMIM基因,该区域的缺失可能与男性生殖细胞的发育与无精症相关。病例10检出X染色体Xp11.23p11.1约11.4Mb的缺失,该区域含有133个OMIM基因,致病性尚不明确。病例11检出9号染色体9q33.3有约420kb的缺失,该区域包含2个OMIM基因,致病性不明确。14号染色体14q11.2有约434kb的缺失,该区域无OMIM基因。病例12检出多条染色体大于10Mb的缺失,但这些缺失区域包含多个隐性致病基因,临床意义尚不明确,杂合性缺失会增加隐性遗传病的发病风险。病例13检出Y染色体Yq11.223q11.23、Yq11.23区域分别有约1.21Mb、1.35Mb的重复,该重复区域与DGVs数据库不完全重叠,共包含7个OMIM基因,但Decipher、Clinvar 等数据库显示该区域重复的临床意义尚不明确。病例14检出4号染色体4q21.21q22.1区域有约12Mb的杂合性缺失,该区域是否携带与遗传印记相关的基因尚不明确,但包含的隐性致病基因会增加隐性遗传病的发病风险。
对于CNV的临床意义不明确,可能是致病性,也可能是良性,即还没有足够的证据或标准证实其临床意义,其结果的解释很具有挑战性,可通过对双亲的进一步检测来明确,如果该CNV来自其没有表型的双亲,则良性的可能性大,而如果是新发的,则致病的可能性更大。但本研究中意义不明确的病例因为其他原因没有进行双亲的检测。
CMA可在全基因组范围进行高分辨率扫描,不仅能检出染色体不平衡性变异,而且能检出亚显微水平的拷贝数变异。缺点是在正常人类基因组中广泛存在没有临床意义的良性CNV/常见 CNV。CMA目前最大问题是对结果的正确解读,其原因在于该技术能找出约1%的不确定性的拷贝数变异,这会给遗传咨询带来一定的困扰,即使CMA存在上述一些缺点,但CNV检测仍具有显著优势,其临床应用价值不可低估[19]。由于正常人CNVs数据库还不完善导致部分检测到的CNVs临床意义判断困难,所以本研究中有6例患者目前仅能判断意义不明CNVs。另外2例缺失区域包含着丝粒位置且探针较少,可能为良性。
CNVs相关的微缺失或微重复是不明原因MR/DD的主要病因,但是如何发现和评估罕见致病性CNVs已成为目前一个面临挑战的重要任务,评估因素主要包括:①是否在正常人群中有报道;②是否为新生CNVs;③CNVs长度是否足够大;④是否在数据库中有报道,且携带者具有RR/DD表型。这些CNVs无法被染色体核型分析所识别,CMA可提高对不明原因MR/DD患儿的分子病因诊断水平,对进一步研究MR/DD病因机制具有重要意义,也可为患儿预后和家庭再发风险评估进行指导。
