应用高通量测序技术分析成品甜芽菜微生物多样性
2019-05-22白光剑邹伟李豪张静
白光剑,邹伟,李豪,张静
(四川理工学院 生物工程学院,四川 宜宾 644000)
芽菜是一种风味独特的传统酱腌菜,在我国川南地区较为盛行[1]。其生产流程包括:第一步,将新鲜的芥菜切成细条状并晒干;第二步,将晒干的芥菜条清洗、加盐混匀并放入容器中发酵1周左右,然后将盐去掉,继续晒干芥菜条;第三步,将芥菜放入桶内,同时添加胡椒、茴香、姜、葱等调料,室温密封发酵6~12个月即成。芽菜可分为甜芽菜和咸芽菜两种。第三步发酵前加入红糖后得到的芽菜属于甜芽菜,不加红糖发酵得到的是咸芽菜。目前宜宾芽菜主要通过自然发酵生产,这一过程中大量环境微生物为宜宾芽菜独特的风味物质形成起着关键作用。国内对芽菜的研究主要集中在芽菜中重要微生物的分离筛选和芽菜风味物质的检测[2-6]。本文主要通过高通量测序技术研究成品芽菜中微生物群落多样性,确定甜芽菜中重要的微生物,为后续工作中加强宜宾芽菜微生物资源的开发提供一定研究基础。
1 材料与方法
1.1 实验材料
实验样品取自自贡市某农贸市场,所有分析样品均为无菌采样,收集3组样品后立即用4~6 ℃的便携式冷藏箱2 h内运到实验室,贮存在冰箱待用。
1.2 样品DNA提取与PCR扩增
样品DNA的提取采用土壤基因组DNA快速提取试剂盒升级版50次(离心柱型),购于BioTeke公司,然后用核酸蛋白仪检测DNA 的浓度和纯度。真菌18S V5-V7区域扩增通用引物:SSU0817F(5′-TTAGCATGGAATAATRRAATAGGA-3′)、1196R(5′-TCTGGACCTGGTGAGTTTCC-3′)。真菌 PCR(50 μL)扩增体系:dNTP Mixture 4 μL,10×PCR Buffer(Mg2+plus)5 μL,正向和反向引物各1 μL,模板 DNA 5 μL,Ex Taq 酶 0.25 μL,ddH2O补足至50 μL,混合均匀。真菌 PCR扩增程序:98 ℃ 3 min;98 ℃ 45 s,53 ℃ 30 s,72 ℃ 45 s,反应共进行30个循环;最后72 ℃ 8 min。细菌 16S rRNA V4 扩增通用引物:16S 338F(5′-ACTCCTACGGAGGCAGCAG-3′)、16S 806R(5′-GGACTACHVGGGTWTCTAAT-3′)。细菌 PCR(50 μL)扩增体系:dNTP Mixture 4 μL,10×PCR Buffer(Mg2+plus) 5 μL,正向和反向引物各 1 μL,模板 DNA 5 μL,Ex Taq 酶 0.25 μL,ddH2O补足至50 μL,充分混匀。细菌PCR扩增程序:95 ℃ 3 min;95 ℃ 45 s,50 ℃ 30 s,72 ℃ 1 min,反应共进行30个循环;最后72 ℃ 5 min。根据 PCR 扩增图谱,扩增后的条带比较清晰明显且条带位置一致,背景干净,DNA 浓度达到了扩增要求,可直接用于后续分析。委托上海美吉生物医药科技有限公司进行Illumina MiSeq高通量测序。
1.3 测序数据优化处理
保证分析结果的准确性,运用Qiime进行序列过滤[7],数据过滤标准为:去除5′端引物错配碱基数>1的序列;去除含有N(模糊碱基)的序列;去除含有连续相同碱基数>8的序列;去除长度≤150 bp的序列;去除嵌合体序列。运用 Mothur软件中的Uchime方法去除嵌合体序列,得到最终用于后续分析的优质序列[8]。
1.4 OTU聚类分析及注释
聚类分析主要通过在Qiime中调用Uclust的方法完成,OUT注释主要通过在Qiime中调用Blast的方法对序列数据库进行比对,OUT精简的方法参考文献[9]。
1.5 多样性及群落结构分析
种群丰富度指数Chao指数和ACE指数,群落多样性指数Shannon指数和Simpson指数,主要通过软件Mothur中的summary.single命令计算获得。不同分类水平上(门、属)的物种丰度表和丰度图主要通过Qiime软件完成。
1.6 稀释曲线制备
稀释性曲线(rarefaction curve)采用对测序序列进行随机抽样的方法,以抽到的序列数与它们所能代表OTU的数目构建。使用97%相似度的OTU利用mothur做rarefaction分析,利用R语言工具制作稀释性曲线。
2 结果与分析
2.1 序列长度分布
测序结果显示:本研究3个芽菜样品所测得的细菌Reads共71140条,分布在401~460 bp长度范围内,平均长度为447.5 bp,长度在441~460 bp范围内占89.38%,有63854条;3个样品所测得的真菌Reads共60900条,分布在261~420 bp长度范围内,平均长度为402.26 bp,长度在401~420 bp范围内占93.18%,有56746条。从序列长度的分布来看,与细菌16S rDNA-V4区序列大致吻合。
2.2 实验样品中微生物群落多样性结果
利用高通量测序技术,经序列过滤和去除嵌合体序列,3个芽菜样品最终得到用于后续数据分析的细菌有效序列65797条,有效率为92.49%;真菌的有效序列54750,有效率为89.90%。本次研究3个样品共产生细菌OTU数609个,每个样品203个,细菌群落分布在7个门、15个纲、31个目、63个科、114个属,见表1。3个样品共产生真菌OTU数109个,样品1和样品3各36个,样品2 OTU数37个,其群落分布在7个门、16个纲、24个目、30个科、28个属中,见表2。
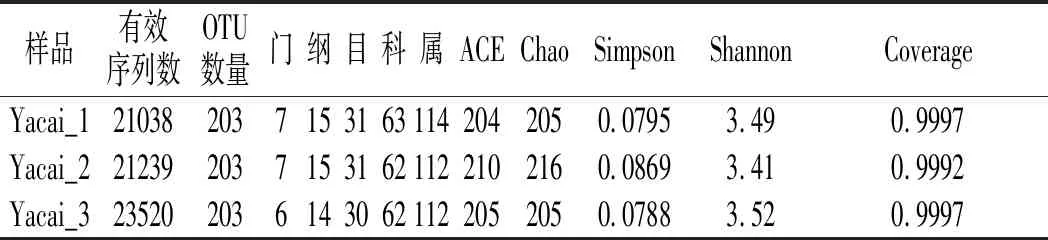
表1 细菌16S rDNA序列丰富度及多样性Table 1 Richness and diversity of bacterial 16S rDNA sequence

表2 真菌18S rDNA序列丰富度及多样性Table 2 Richness and diversity of fungal 18S rDNA sequence
2.3 多样性指数分析
群落生态学中研究微生物多样性,通过单样品的多样性分析(Alpha多样性)可以反映微生物群落的丰度和多样性,包括一系列统计学分析指数估计环境群落的物种丰度和多样性。有用于计算菌群丰度(community richness)的指数Chao、ACE,用于计算菌群多样性(community diversity)的指数Simpson、Shannon,以及代表测序深度的指数Coverage。其中Chao或ACE指数越大,说明群落丰富度越高;Shannon值越大,说明群落多样性越高;Simpson指数值越大,说明群落多样性越低;Coverage值越高,则样本中序列被测出的概率越高。
由表1和表2可知,细菌的Chao和ACE指数均大于真菌,说明成品甜芽菜中细菌群落丰富度大于真菌群落;从Simpson和Shannon指数来看,细菌群落的多样性也高于真菌;两者Coverage指数都非常接近1,表明本次测序结果能代表样本中微生物的真实情况。
2.4 微生物多样性稀释性曲线
综合数据和稀释曲线分析来看(见图1),在较少的测序数范围内,真菌和细菌均有大量的OTU产生,细菌较真菌有更多的OTU。各样本所代表的曲线斜率,在测序数5000左右骤然下降,真菌的稀释曲线有少量新的OTU产生,细菌OTU变化很小,随着测序深度增加,真菌和细菌的稀释曲线斜率趋于平稳。说明测序数据量合理,能有效地反映芽菜中微生物的丰富度和多样性。


图1 芽菜样品中微生物变化稀释曲线 Fig.1 Microbial variation dilution curves of samples
注:A为细菌变化稀释曲线图;B为真菌变化稀释曲线。
2.5 微生物丰度分析
由于通常测序分析,芽菜样品能检测出大量微生物种类,但许多物种含量稀少,不能与含量多的物种明显体现在同一个图中,所以将物种的丰度大于1%作为划分依据,以相对丰度为纵坐标,绘制柱形图。从细菌门的分类水平来看,3个样品所得结果基本相同,共检测出7个细菌门,分别是放线菌门 (Actinobacteria)、拟杆菌门 (Bacteroidetes)、蓝细菌(Cyanobacteria)、厚壁菌门(Firmicutes)、梭杆菌门(Fusobacteria)、变形菌门 (Proteobacteria)、无壁菌门(Tenericutes)。其中优势细菌有变形菌门(49.1%)、厚壁菌门(40.9%)、放线菌门(5.1%)、拟杆菌门(2.6%)、蓝细菌(2.6%)(所得丰度值百分比取3个样品的平均值,下同),其他细菌菌门丰度值均小于1%,(见图2A)。从细菌属的分类水平来看,共检测出114个属,按其丰度值排序,分别是盐单胞菌属(Halomonas,27.2%)、芽孢杆菌属(Bacillus,11.9%)、肠球菌属(Enterococcus,6.8%)、Lactococcus,96.2%)、假单胞菌属(Pseudomonas,5%)、Enterobacteriaceae_unclassified(4.8%)、盐厌氧菌属(Halanaerobium,4.5%)、欧文氏菌属(Erwinia,2.6%)、棒状杆菌属(Corynebacterium_1,2.5%)、魏斯氏菌属(Weissella,2.2%)、乳酸杆菌属(Lactobacillus,2.2%)、Cyanobacteria_norank(2.1%)、拉恩菌属(Rahnella,1.4%)、葡萄球菌属(Staphylococcus,1.4%)、Alkaliphilus(1.4%)、Acinetobacter(1.0%),其他菌群丰度小于1%,占总菌群丰度的15.2%(见图2B)。
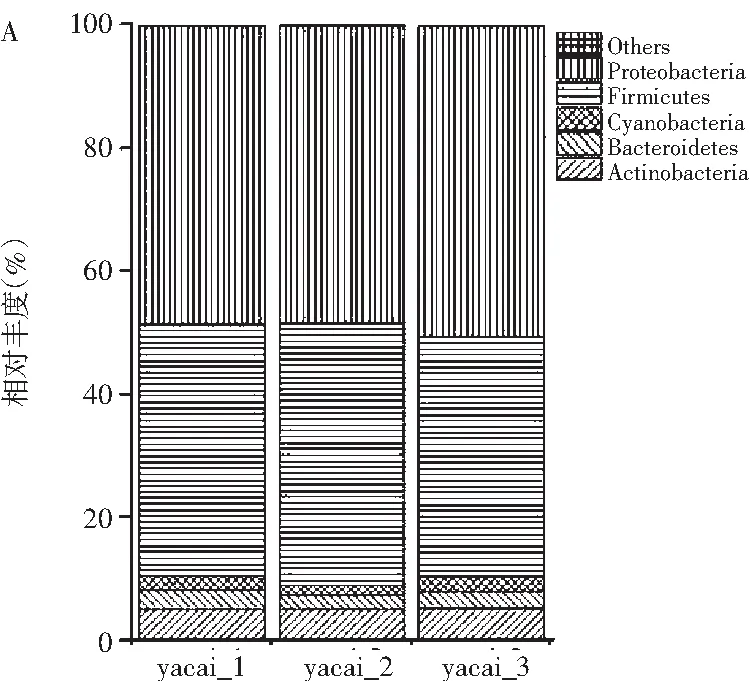
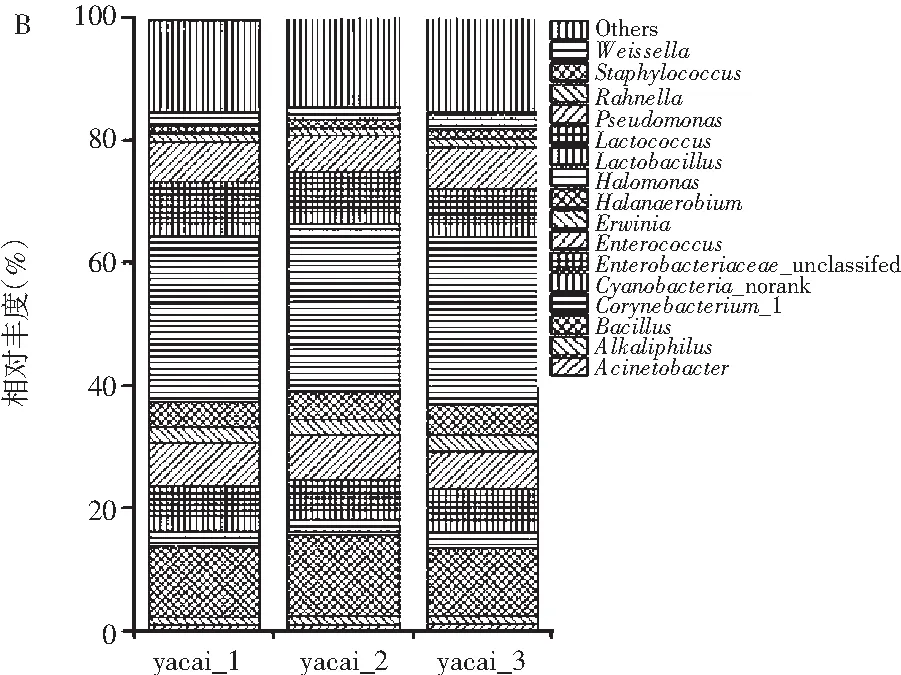
图2 成品甜芽菜细菌群落门(A)和属(B)水平相对丰度Fig.2 Relative abundance of bacteria in the finished product of sweet sprouts at phylum(A)and genus(B)levels
从真菌门的分类水平来看,3个样品共检测出6个门的真菌:Arthropoda、子囊菌门(Ascomycota)、担子菌门(Basidiomycota)、Ciliophora、Phragmoplastophyta、结合菌门(Zygomycota),其中占优势的门是Ascomycota(63.7%),Basidiomycota(36.1%),其他 4个菌门丰度所占百分比之和约占0.2%(见图3A)。从真菌属的分类水平来看,共检测到28个属,按其丰度排序分别是Saccharomycetales_unclassified(28.5%),样品2的略高于其他两个样品,德巴利酵母属(Debaryomyces,15.8%),Filobasidiaceae_norank(14.1%),Cladosporium(7.1%),Itersonilia(6.6%),Incertae_Sedis_Incertae_sedis(6.1%),Leucosporidiales_norank(5.3%),Pleosporaceae_unclassified(5.2%),核盘菌属(Sclerotinia,5.0%),Tremellales_norank(3.2%),其他菌群丰度均小于1%,占总菌群丰度的3.1%(见图3B)。
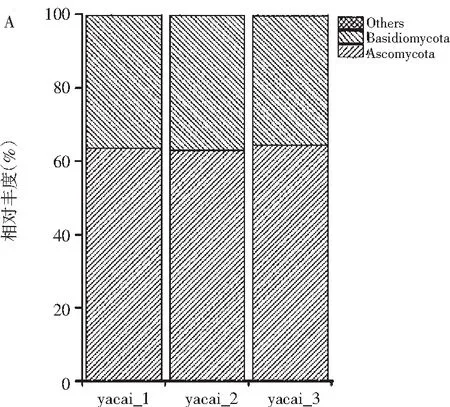

图3 成品甜芽菜真菌群落门(A)和属(B)水平相对丰度Fig.3 Relative abundance of fungi in the finished product of sweet sprouts at phylum(A)and genus(B)levels
3 讨论
本文采用Miseq高通量测序技术对成品甜芽菜中细菌和真菌多样性进行研究。对细菌的测序显示,共得到65797条有效序列,聚类分析共产生609个OTU,3个样品较为平均,各有203个,分属7个门114个属内,其中从门水平来看Proteobacteria(49.1%),Firmicute(40.9%)具有较大优势;从属水平来看,盐单胞菌属(Halomonas,27.2%)的丰度最高,芽孢杆菌属(Bacillus,11.9%)次之。与前期报道芽菜发酵过程细菌多样性相比[10],Bacillus和Pseudomonas均为优势菌株,但成品芽菜中的Halomonas,Enterococcus,Lactococcus在发酵过程中优势不明显。发酵过程中的优势菌株如Flavobacteriaceae,Sphingobacterium,Sphingomonas,Acidovorax,Chromohalobacter,Arthrobacter在成品甜芽菜中优势并不明显。真菌的测序共得到有效序列54750条,聚类分析共产生109个OTU,分属7个门28个属。从门水平来看,Ascomycota(63.7%),Basidiomycota(36.1%)具有绝对优势,其他真菌门丰度较小。从属水平结果看Saccharomycetales_unclassified(28.5%),Debaryomyces(15.8%),Filobasidiaceae_norank(14.1%)3个菌属较为优势。与芽菜发酵过程相比[11],发酵150天时Debaryomyces(62%)相比优势下降。可以看出即便是与发酵末期相比,芽菜中的细菌和真菌群落结构和丰度都发生了不小的变化。
目前,已有学者对芽菜发酵过程以及成品芽菜的风味物质成分进行了系统研究[12]。成品甜芽菜的主要风味成分为丁酸、E-11-棕榈酸乙酯、2,3-丁二醇、2,6,10-三甲基十四烷、芥子酸、1-十八烷烯、1H-吲哚-3-乙腈。解析芽菜中微生物群落组成与其风味物质产生之间的关系不仅有助于理解芽菜的风味物质形成机制,同时也有助于进一步控制芽菜质量,但相关研究较少,将是未来研究的热点。
