利用CRISPR/Cas9和piggyBac实现果蝇基因组无缝编辑
2019-05-21王珏黄娟许蕊
王珏,黄娟,许蕊
利用CRISPR/Cas9和实现果蝇基因组无缝编辑
王珏,黄娟,许蕊
南京医科大学基础医学院医学遗传学系,南京 211166
CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9)是第三代基因组编辑技术。在sgRNA引导下,Cas9核酸内切酶作用于特定基因组序列,产生DNA双链断裂(double-stranded breaks, DSBs),利用同源定向修复(homology-directed repair, HDR)可实现对靶基因的特异性基因敲除(knock-out)或敲入(knock-in)。传统的技术方案将CRISPR/Cas9技术与Cre/loxP或FLP/FRT系统联用,可实现高效的基因打靶,也易于移除打靶过程中引入的筛选标记。然而,筛选标记移除过程中会在基因组中残留34个碱基的标签序列。因此,对基因组进行精确编辑的同时不引入无关序列仍有一定难度。在人工诱导多能干细胞(induced pluripotent stem cells, iPSCs)的基因组编辑中,CRISPR/Cas9技术和转座酶联用的两步法策略能够实现这一目标:首先运用CRISPR/Cas9技术,利用同源定向修复原理引入基因突变及筛选标记,然后利用转座酶将筛选标记精确移除。借鉴该方法的技术原理,本研究对果蝇()基因进行了无缝编辑(seamless genome editing),成功将该基因第18外显子上第21位的酪氨酸(tyrosine, Y)突变为半胱氨酸(cysteine, C),且测序结果显示基因组中除设计位点之外并无其他外源序列残留。CRISPR/ Cas9技术和转座酶联用策略为果蝇基因组的精确编辑提供了更多选择。
果蝇;无缝基因组编辑;CRISPR/Cas9;
利用现代生物学技术获得基因突变体,并检测基因突变造成的影响是研究基因功能的重要手段。CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9)技术的出现和应用,为实现基因打靶提供了简单高效的方法[1~3]。在sgRNA引导下,Cas9核酸内切酶可作用于特定基因序列,产生DNA双链断裂(double-stranded breaks, DSBs),诱发内源性非同源末端连接(non-homologous end joining, NHEJ)或同源定向修复(homology-directed repair, HDR)。
非同源末端连接可导致修复位点产生小片段的DNA插入或缺失,从而造成目标基因突变。但是,这类突变具有一定的随机性,无法实现对目标基因的特定编辑。如果在DNA双链断裂修复过程中提供外源模板,则在同源定向修复启动时可将模板中的特异性突变引入基因组中,从而实现对目标基因的特定编辑。然而,同源定向修复的效率远远低于非同源末端连接[4]。为了高效筛选到携带特异性突变的转基因果蝇()品系,需同时引入筛选标记,以便判断是否发生了同源定向修复。筛选标记的移除通常利用Cre/loxP或FLP/FRT系统进行,但它们都会在基因组中遗留34个碱基的标签序列[5]。如何在对基因组进行精确编辑的同时不引入无关序列,即实现基因组无缝编辑(seamless genome editing)仍有一定难度。
转座子系统来源于鳞翅目昆虫,其转座酶识别DNA两端的特定反向末端重复序列(inverted terminal repeat sequences, ITRs),并将其插入染色体中TTAA位点[6,7]。该系统简单高效,早期用在果蝇、蚊子()等昆虫中获得转基因品系[8,9]。近年来研究发现,转座子系统在哺乳动物细胞中也具有很高的转座活性[10,11]。除了能够将外源基因转入基因组,转座酶还能够将带有特定ITRs的DNA序列从基因组中精确移除。该系统与CRISPR/Cas9技术联用,能够用来移除在人诱导多能干细胞(induced pluripotent stem cells, iPSCs)基因组编辑中引入的药物筛选标记或核酸酶,且不遗留任何外源核酸序列,实现基因组无缝编辑[12,13]。
果蝇基因位于2号染色体左臂,其编码的Ca-α1D蛋白是L型电压门控钙离子通道(voltage- gating calcium channel, VGCC)的重要组成亚基,参与肌肉收缩、神经重塑、上皮液转运等一系列重要的生命过程[14~17]。研究Ca-α1D蛋白,尤其是其特定结构域的功能对揭示电压门控钙离子通道的生理功能具有重要意义。
本研究以果蝇为材料,利用CRISPR/Cas9技术和转座子系统将基因第18个外显子上第21位酪氨酸(tyrosine, Y)突变为半胱氨酸(cysteine, C),且没有引入任何外源序列。本研究应用的基因打靶策略为果蝇基因组的精确编辑提供了更多选择。
1 材料与方法
1.1 材料
野生型果蝇w、平衡染色体品系和转座酶表达品系BL#8284均来自于Bloomington果蝇库,BL#8284果蝇品系2号染色体携带并与标记相偶联,可内源性表达转座酶。Nos-Cas9(3rdchr)果蝇品系为本实验室果蝇库所有,该品系3号染色体attP2位置插入了基因,胚胎细胞能够内源性表达Cas9核酸内切酶。U6b-sgRNA-short载体由清华大学医学院倪建泉实验室构建[18],pGX-attP载体为本实验室构建[19]。
1.2 果蝇基因组测序
在Flybase数据库中查询基因序列信息,设计引物对Nos-Cas9(3rdchr)果蝇品系基因第18外显子(长度为108 bp)上下游各约300 bp进行PCR扩增及测序(引物序列见表1),扩增片段大小为700 bp,经测序,结果表明序列与Flybase数据库中相同。这一步骤的目的是确保sgRNA能顺利与基因组中相应序列匹配,并正确引导Cas9切割靶序列。由于基因内含子区域常存在比较大的变异性,当sgRNA序列位于内含子区域时,这一步尤为重要。
1.3 sgRNA质粒构建
首先在目标突变位点附近寻找带有PAM (protospacer adjacent motif)的序列作为sgRNA序列,将对应序列(5′-CACCAACATGATATTACCAA-3′)在果蝇CRISPR网站(http://flycrispr.molbio.wisc.edu)上检测,没有发现可能的脱靶序列。根据sgRNA序列设计2条5′端分别带有TTCG和AAAC序列的互补引物FW2和RV2 (序列见表1)。将2种引物分别配制成100 μmol/L溶液,各取10 μL,加入2.5 μL 10×的T4 DNA连接酶缓冲液,并补水2.5 μL至终体积25 μL,在PCR仪上复性(复性条件为100℃ 10 min,之后每分钟温度降1℃至最终温度25℃)形成5′末端带有粘性末端的双链DNA片段,通过T4 DNA连接酶(NEB公司,美国)连接在经Ⅰ酶切后回收的载体U6b-sgRNA-short上,获得sgRNA表达质粒,方法见参考文献[20]。
1.4 供体DNA质粒构建
将pGX-attP载体(载体详情见参考文献[19])上两个多克隆位点之间的(从65Ⅰ位点至Ⅰ位点)替换为两端带有转座酶识别位点ITRs的标记基因,获得载体pGX- DsRed。在目标突变位点附近寻找TTAA位点作为标记基因插入位置,在此序列前后各约1.2 kb和1.1 kb分别设计5′同源臂和3′同源臂,同源臂扩增使用的PCR酶为Phanta高保真酶(南京诺唯赞公司)。扩增5′同源臂的引物为FW3、RV3、FW4和RV4(序列见表1),为了引入突变碱基,本研究将5′同源臂分成2部分进行扩增。由于供体DNA质粒中包含sgRNA序列,为防止供体质粒注入果蝇胚胎后被Cas9切割,本研究对这部分序列中的3个氨基酸进行了同义突变(GGT→GGA,AAT→AAC,ATC→ ATT),同时扩增过程中还在FW3引物中引入了点突变(TAT→TGT),编码氨基酸由酪氨酸变成半胱氨酸。3′同源臂扩增引物为FW4和RV4(序列见表1)。为保证标记在后续实验过程中顺利移除,5′同源臂的3′端和3′同源臂的5′端均带有TTAA序列,这样经转座酶处理后才能得到无缝效果。扩增得到的片段使用多片段同源重组酶(南京诺唯赞公司)进行连接。

表1 引物序列信息
斜体下划线分别为酶切位点RⅠ和65Ⅰ序列,仅下划线为同义突变碱基,粗体下划线为点突变碱基,粗体为TTAA序列。
1.5 基因敲入果蝇突变体的获得
取sgRNA表达质粒5 μg、供体DNA质粒10 μg,混合后用酚/氯仿抽提纯化,并用20 μL注射缓冲液(5 mmol/L氯化钾,0.1 mmol/L磷酸钠,pH 6.8)溶解质粒,使sgRNA质粒和供体DNA质粒终浓度分别为250 ng/μL和500 ng/μL。收集Nos-Cas9(3rdchr)果蝇胚胎进行显微注射,孵化出的雌蝇和雄蝇分别与w的雄蝇和处女蝇杂交,筛选后代携带红色荧光标记的果蝇,并与平衡染色体品系杂交。对筛选到的转基因果蝇进行鉴定时,分别在基因组和插入的标记基因上设计引物用于跨同源臂PCR鉴定。跨5′同源臂PCR产物大小约为1.8 kb,上游引物FW6位于基因组5′同源臂上游,下游引物RV6位于基因近5′端;跨3′同源臂PCR产物大小约为1.2 kb,上游引物FW7位于基因组3′同源臂上游,下游引物RV7位于基因近3′端。引物序列见表1。经1%琼脂糖凝胶电泳检测确认扩增条带大小是否正确。
1.6 筛选标记基因的切除
将鉴定正确的转基因果蝇品系与表达转座酶的果蝇品系BL#8284杂交,在杂交后代中挑选基因型为[DsRed]*的杂合子,该基因型的杂合子部分细胞中的标记被转座酶切除而显示出镶嵌表型,可通过观察复眼的镶嵌型红色荧光表型挑选该基因型的果蝇,并根据复眼中红色荧光存留比例估算大致移除效率。将该杂合子与平衡染色体品系杂交,在后代中挑选不带标记且带有标记的果蝇,并与平衡染色体品系杂交,并使其纯合。提取纯合果蝇基因组DNA,对基因第18个外显子附近进行PCR扩增及测序,确定纯合品系是否携带点突变。
2 结果与分析
2.1 CG4894敲入突变体的获得及鉴定
本研究通过查询Flybase数据库获得基因序列,发现目标氨基酸—酪氨酸位于第18外显子第21位,此外显子为转录本C、E、H和J共有,其序列见图1A (大写粗体部分)。本研究通过PCR扩增及测序,验证了第18外显子上下游约300 bp的序列与数据库中查询序列相同,在目标氨基酸附近选取合适的sgRNA序列和TTAA序列。
在构建供体DNA质粒时,本研究以目标突变位点附近的TTAA序列为界,分别选取TTAA上游1.2 kb序列作为5′同源臂,TTAA下游1.1 kb序列作为3′同源臂。由于5′同源臂包含了sgRNA序列,为防止Cas9剪切,本研究将sgRNA序列中3个氨基酸进行了同义突变。5′同源臂分为2个部分进行扩增,并通过引物引入sgRNA中的同义突变和21位氨基酸的点突变(图1B),成功进行同源重组后两端带有ITRs的基因被插入基因组TTAA位置。后续实验中,转座酶可将2个TTAA之间的标记移除,恢复为原基因组中的1个TTAA序列,实现基因组的无缝编辑。
将sgRNA质粒和供体DNA质粒混合物注入约1000个Nos-Cas9(3rdchr)果蝇胚胎中,最终得到257只雄蝇和240只雌蝇,注射成活率为49.7%。这些雄蝇和雌蝇分别和w处女蝇、雄蝇进行杂交,后代经过红色荧光筛选,获得3个携带筛选标记的果蝇品系。杂交实验表明,3个品系中标记均位于目标基因所在的2号染色体上,其编号分别为1-1、5-1和5-2。此外,如果发生了同源重组,利用FW6、RV6引物扩增可得到1.8 kb大小、跨5′同源臂的PCR产物,利用FW7、RV7引物扩增可得到1.2 kb大小、跨3′同源臂的PCR产物,电泳结果显示3个品系的基因组DNA均能扩增得到预期大小的条带,证实本研究已成功构建了敲入突变体(图1C)。
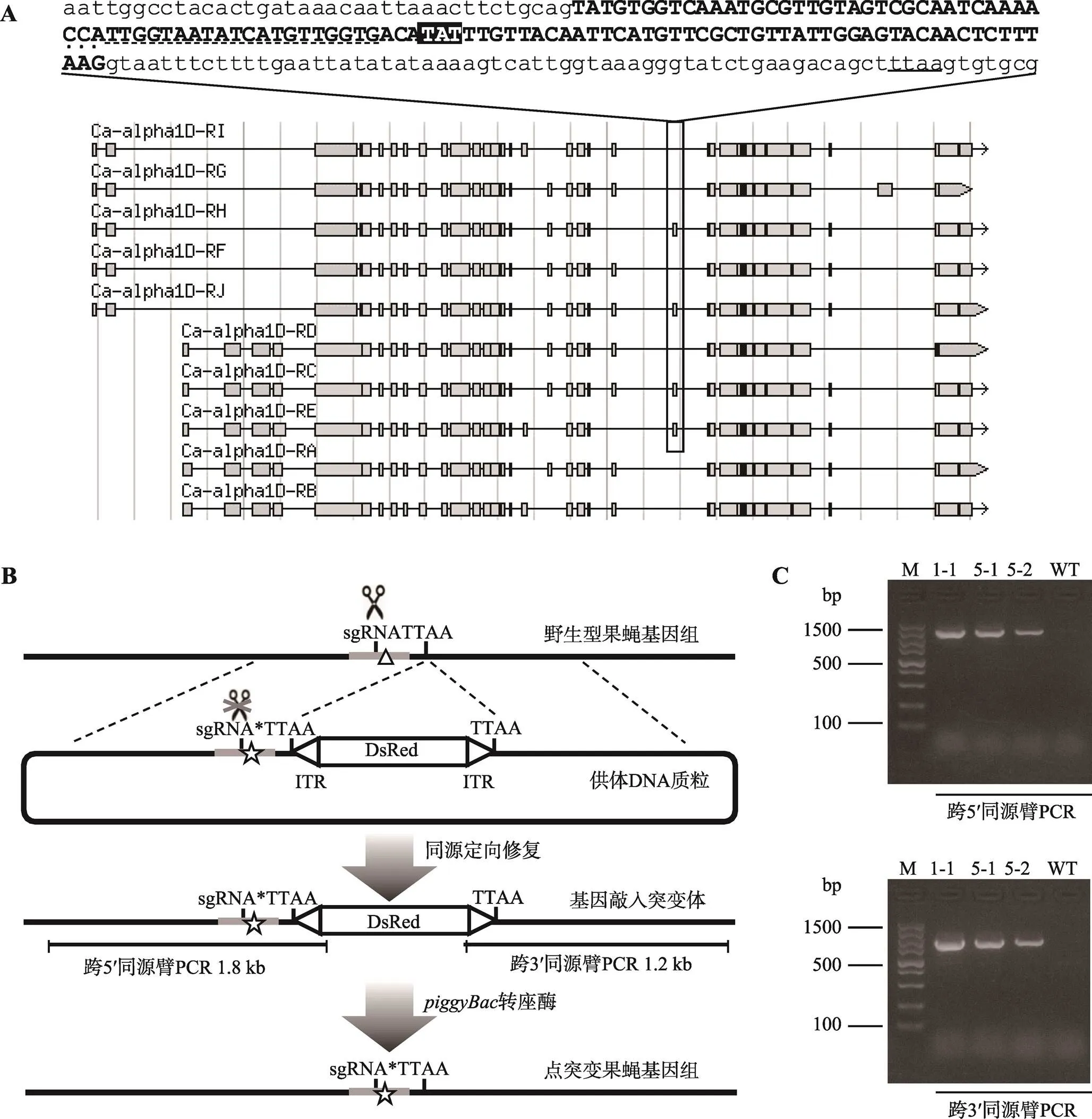
图1 基因敲入突变体的设计及鉴定
A:基因转录本第18外显子示意图。下方为基因转录本示意图(引自Flybase数据库),方框标记表示部分转录本C、E、H和J共有的第18外显子,上方为这一外显子及其上下游序列,阴影部分为目标突变位点,实线下划线为TTAA序列,虚线下划线为sgRNA序列,加点CCA为PAM序列;B:基因点突变体构建示意图。三角形标记为基因组中目标点突变位置,五角星标记为供体DNA质粒携带的点突变,sgRNA*表示供体DNA质粒上的sgRNA携带同义突变;C:基因敲入果蝇跨同源臂PCR鉴定结果。M:100 bp DNA maker;1-1、5-1、5-2为携带筛选标记的转基因果蝇品系;WT:野生型果蝇。
2.2 DsRed荧光标记的去除和鉴定
为了移除上述基因敲入品系中的筛选标记,本研究通过杂交方式引入转座酶。BL#8284果蝇品系2号染色体上带有由hsp70启动子驱动表达的转座酶,将这一品系与带有标记的转基因品系杂交,后代基因型为[DsRed]*的杂合体表现出镶嵌表型,即部分细胞中标记被转座酶切除,且红色荧光存留比例取决于的表达和剪切效率。本研究杂合体果蝇复眼中约有50%的小眼无红色荧光,据此估算转座酶的切除效率大概为50%左右。将镶嵌表型果蝇与平衡系果蝇杂交,后代出现的无红色荧光果蝇(图2A)与平衡染色体品系杂交,并使其纯合。3个品系中携带筛选标记的转基因果蝇1-1、5-1和5-2经过杂交,均获得了不携带荧光标记的白眼点突变纯合果蝇,这些品系未出现明显表型。
理论上,同源重组可以发生在同源臂的任意位置。在本研究中,如果同源重组发生在突变位点和TTAA之间,则突变不被引入基因组。因此,本研究对获得的白眼点突变纯合果蝇品系进行了测序,验证突变是否引入。测序结果表明,3个品系中均存在Y→C突变及sgRNA部分的同义突变(图2B),且TTAA位点附近没有引入任何突变和外源序列(图2C)。
综上所述,本研究成功在果蝇基因中引入了点突变,并且没有任何外源序列遗留,证实了CRISPR/Cas9和转座子系统联用策略用于果蝇基因组精确编辑的可行性。
3 讨论
基因组编辑技术能够精确地实现生物体基因组定点突变、插入或敲除,人为改变生物遗传信息。20世纪末人们就开始对基因编辑技术进行探索,从第一代DNA核酸酶编辑系统ZFN (zinc finger nuclease)、第二代TALEN (transcription activator-like effector nuclease)到第三代CRISPR/Cas9系统,基因编辑效率不断提高、成本逐渐降低,应用范围不断扩大[21]。近几年CRISPR/Cas9技术的迅速发展,及其在各种模式生物中的广泛应用,使得突变体的获得变得更加简单高效。与此同时,人们对突变体的特异性也提出了越来越高的要求。如前所述,NHEJ容易引起随机插入和缺失,它导致的突变带有很大的随机性,想要获得“设计”的突变必须通过同源定向修复。但在这一策略中,提高突变体筛选的效率和不引入无关序列就成了一对矛盾,这一矛盾在果蝇中尤为突出。由于果蝇体型较小,无法使用部分身体组织提取DNA,只能在子代产出之后再将亲本处死,提取DNA进行基因型鉴定,因此在果蝇基因编辑过程中多引入各种筛选标记,通过对后代进行表型筛选提高突变体的筛选效率。

图2 标记基因的去除和点突变体的测序鉴定
A:去除标记基因的杂交流程。星号表示部分标记基因移除;B:野生型果蝇和基因点突变体在目标突变位点处的序列比较。五角星标记表示目标突变位点,星号表示同义突变位点;C:野生型果蝇和基因点突变体在TTAA位点处的序列比较。WT:野生型果蝇;1-1、5-1、5-2为白眼点突变纯合果蝇品系。

图3 3种基因组编辑策略的比较
A:介导的基因组无缝编辑。标记基因两端带有转座酶特异识别序列(ITRs),CRISPR/Cas9介导的同源重组使其随着突变插入染色体TTAA位点,后经转座酶切除,只在染色体中保留突变;B:单链退火(single-strand annealing, SSA)介导的DNA双链断裂修复。两条同源臂在基因组中有重叠,因此第一次同源重组导致染色体上携带重复片段,且重复片段位于标记基因两端,再次利用CRISPR/Cas9制造染色体断裂可以诱导染色体内同源重组的发生,将标记基因去除;C:通过两步同源重组实现的无缝基因置换。第一次同源重组将目标基因替换为标记基因,第二次同源重组将标记基因替换为带有突变的目标基因。其中,目标基因两端可以携带不同于基因组剪切所用的sgRNA序列,以保证Cas9不会作用于同源臂载体。图根据文献[13,22,23]修改绘制,三角形标记表示基因组中目标点突变位置,五角星标记表示供体DNA质粒携带的点突变。
为了精确去除筛选标记,在人诱导多能干细胞的基因组编辑中,转座酶被尝试用于去除基因编辑过程中引入的药物筛选标记,以实现基因无缝编辑,并获得了成功(图3A)。除此以外,还有2种策略已被报道。单链退火(single-strand annealing, SSA)介导的DNA双链断裂修复(SSA-mediated DNA double-strand break repair)[22]在HEK293T细胞基因编辑过程中得到应用,该策略是在同源重组阶段通过带有重叠的同源臂引入染色体内重复,然后介导染色体内同源重组将标记基因去除(图3B)。两步同源重组实现的无缝基因置换[23]被用于果蝇基因的编辑,该策略利用第一次同源重组将目标基因替换为标记基因,再利用第二次同源重组将标记基因替换为突变后基因(图3C)。但这两种方法与转座酶法相比,筛选标记的去除都需通过同源重组,因此稍显复杂。
转座子系统简单高效,不需二次基因打靶就能将标记基因精确切除。而且,由于这一系统早就用于果蝇的基因编辑,果蝇中已发展了一系列相关工具,如本研究中用到的内源表达转座酶的果蝇品系。这一品系使得标记基因的去除可以通过简单的杂交进行,而不像其他系统,需要通过注射或其他复杂操作将转座酶引入突变体。当然,该系统对基因序列有一定要求,标记基因需要插入内源的TTAA序列,如果其距离目标突变位点较远则会降低突变引入的效率。理论上,TTAA在基因组中出现的概率为1/256,由于TTAA只需在目标区段附近即可,因而不难找到。
综上所述,CRISPR/Cas9和转座子系统联用策略在果蝇中的可行性为果蝇基因组的精确编辑提供了更多选择。
[1] Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems., 2013, 339(6121): 819–823.
[2] Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity., 2012, 337(6096): 816–821.
[3] Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineeringCas9., 2013, 339(6121): 823–826.
[4] Miyaoka Y, Berman JR, Cooper SB, Mayerl SJ, Chan AH, Zhang B, Karlin-Neumann GA, Conklin BR. Systematic quantification of HDR and NHEJ reveals effects of locus, nuclease, and cell type on genome-editing., 2016, 6: 23549.
[5] van der Weyden L, Adams DJ, Bradley A. Tools for targeted manipulation of the mouse genome., 2002, 11(3): 133–164.
[6] Cary LC, Goebel M, Corsaro BG, Wang HG, Rosen E, Fraser MJ. Transposon mutagenesis of baculoviruses: analysis of Trichoplusia ni transposon IFP2 insertions within the FP-locus of nuclear polyhedrosis viruses., 1989, 172(1): 156–169.
[7] Wang HH, Fraser MJ, Cary LC. Transposon mutagenesis of baculoviruses: analysis of TFP3 lepidopteran transposon insertions at the FP locus of nuclear polyhedrosis viruses., 1989, 81(1): 97–108.
[8] Handler AM, Harrell RA 2nd. Germline transformation ofwith thetransposon vector., 1999, 8(4): 449–457.
[9] Lobo N, Li X, Fraser MJ, Jr. Transposition of theelement in embryos of, aedes aegypti and trichoplusia ni., 1999, 261(4–5): 803–810.
[10] Ding S, Wu X, Li G, Han M, Zhuang Y, Xu T. Efficient transposition of the(PB) transposon in mammalian cells and mice., 2005, 122(3): 473–483.
[11] Wu SC, Meir YJ, Coates CJ, Handler AM, Pelczar P, Moisyadi S, Kaminski JM.is a flexible and highly active transposon as compared to sleeping beauty, Tol2, and Mos1 in mammalian cells., 2006, 103(41): 15008–15013.
[12] Wang G, Yang L, Grishin D, Rios X, Ye LY, Hu Y, Li K, Zhang D, Church GM, Pu WT. Efficient, footprint-free human iPSC genome editing by consolidation of Cas9/CRISPR andtechnologies., 2017, 12(1): 88–103.
[13] Yusa K. Seamless genome editing in human pluripotent stem cells using custom endonuclease-based gene targeting and thetransposon., 2013, 8(10): 2061–2078.
[14] Zheng W, Feng G, Ren D, Eberl DF, Hannan F, Dubald M, Hall LM. Cloning and characterization of a calcium channel alpha 1 subunit fromwith similarity to the rat brain type D isoform., 1995, 15(2): 1132–1143.
[15] Kanamori T, Kanai MI, Dairyo Y, Yasunaga K, Morikawa RK, Emoto K. Compartmentalized calcium transients trigger dendrite pruning insensory neurons., 2013, 340(6139): 1475–1478.
[16] Hara Y, Koganezawa M, Yamamoto D. The Dmca1D channel mediates Ca(2+) inward currents inembryonic muscles., 2015, 29(2–3): 117–123.
[17] Limpitikul WB, Viswanathan MC, O'Rourke B, Yue DT, Cammarato A. Conservation of cardiac L-type Ca(2+) channels and their regulation in: a novel genetically-pliable channelopathic model., 2018, 119: 64–74.
[18] Ren X, Sun J, Housden BE, Hu Y, Roesel C, Lin S, Liu LP, Yang Z, Mao D, Sun L, Wu Q, Ji JY, Xi J, Mohr SE, Xu J, Perrimon N, Ni JQ. Optimized gene editing technology forusing germ line-specific Cas9., 2013, 110(47): 19012–19017.
[19] Huang J, Zhou W, Dong W, Watson AM, Hong Y. Directed, efficient, and versatile modifications of thegenome by genomic engineering., 2009, 106(20): 8284–8289.
[20] Tang JB, Cao HW, Xu R, Zhang DD, Huang J. Mutant generation of the testis genes and phenotype analyses in., 2018, 40(6): 478–487.唐浚博, 曹浩伟, 许蕊, 张丹丹, 黄娟. 果蝇睾丸基因敲除突变体的构建及表型分析. 遗传, 2018, 40(6): 478–487.
[21] Ren YX, Xiao RD, Lou XM, Fang XD. Research advance and application in the gene therapy of gene editing technologies., 2019, 41(1): 18–27.任云晓, 肖茹丹, 娄晓敏, 方向东. 基因编辑技术及其在基因治疗中的应用. 遗传, 2019, 41(1): 18–27.
[22] Li X, Bai Y, Cheng X, Kalds PGT, Sun B, Wu Y, Lv H, Xu K, Zhang Z. Efficient SSA-mediated precise genome editing using CRISPR/Cas9., 2018, 285(18): 3362–3375.
[23] Lamb AM, Walker EA, Wittkopp PJ. Tools and strategies for scarless allele replacement inusing CRISPR/Cas9., 2017, 11(1): 53–64.
Seamless genome editing inby combining CRISPR/Cas9 andtechnologies
Jue Wang, Juan Huang, Rui Xu
The typeⅡ CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR- associated protein 9) is an efficient RNA-guided genome-editing technique. Guided by sgRNA, the Cas9 endonuclease generates site-specific double-stranded breaks (DSB) at specific site, which is amenable to repair by homology-directed repair (HDR) to generate a designed knock-out or knock-in transgene. In combination with CRISPR/Cas9 and Cre/loxP or FLP/FRT system, efficient gene targeting can be achieved, and meanwhile screening markers introduced can be readily removed except a 34-base pair residual fragment. Thus, difficulties remain in accurate editing of the genome without introducing any extraneous sequences. In human induced pluripotent stem cells (iPSCs), a two-step strategy has been developed using CRISPR/Cas9 and thesystem to establish a seamless genomic editing, in which CRISPR/Cas9 is initially used to introduce mutations along with screening markers by HDR, then the markers are precisely excised bytransposase. Using this strategy, we have successfully transformed the tyrosine to cysteine at position 21 within the 18th exon of thegene in thegenome without introducing any extraneous sequence. Hence, this strategy provides more options for precise and seamless editing of thegenome.
; seamless genome editing; CRISPR/Cas9;
2018-12-27;
2019-01-31
南京医科大学引进人才启动经费项目(编号:2012RC04)资助[Supported by the Start-up Foundation from Nanjing Medical University (No. 2012RC04)]
王珏,本科,专业方向:果蝇基因打靶技术。E-mail: 15651972835@163.com
许蕊,硕士,实验师,研究方向:果蝇基因打靶技术。E-mail: xurui20062624@163.com
10.16288/j.yczz.18-345
2019/3/29 13:36:20
URI: http://kns.cnki.net/kcms/detail/11.1913.R.20190329.1335.002.html
(责任编委: 张雷)
