大卫颗粒的质量标准提高△
2019-05-13李晓王学方张丽先李飞飞宁二娟魏悦
李晓,王学方,张丽先,李飞飞,宁二娟,魏悦
1.河南省生物技术开发中心,河南 郑州 450002;2.河南省植物天然产物开发工程技术研究中心,河南 郑州 450002
大卫颗粒由连翘、黄芩、柴胡、金银花、甘草、紫苏叶6味中药组成,收载于《中药部颁标准》(第十册),具有清热解毒、疏风透表之功效[1]。临床主要用于感冒发烧、头痛、咳嗽、鼻塞流涕、咽喉肿痛等症,研究表明,大卫颗粒治疗小儿病毒性上呼吸道感染总有效率达92.4%,明显优于利巴韦林对照组(79.1%)[2];此外,大卫颗粒对妊娠期急性上呼吸道感染的临床症状有明显缓解、可增强机体免疫力,总有效率为92.31%[3]。
但该制剂现行标准较为落后,只收载了薄层鉴别法鉴别绿原酸和相关检查项目,前期有文献报道了大卫颗粒中绿原酸、黄芩苷等成分的含量测定方法[4-5],但是其全面的质量标准研究未见报道。大卫颗粒已上市多年,临床应用量大[6],尤其是在儿科[7],故有必要加强其质控方法和标准研究,保证其临床疗效与安全性。
为此,本研究对制剂中主要药味连翘、金银花、柴胡等进行薄层色谱(TLC)鉴别,并对其中绿原酸、连翘酯苷A、黄芩苷的含量进行高效液相色谱(HPLC)测定,为其质量标准提高提供参考。
1 材料
1.1 仪器
LC-20AT高效液相色谱仪(日本岛津,CMA-20A系统、LC-20AT二元高压泵、SIL-20A自动进样器、CTO-20A柱温箱、SPD-M20A检测器、LC Soulation工作站);GoodSee-20E薄层色谱成像系统(上海科哲生化科技有限公司);ME204型万分之一分析天平[托利多仪器(上海)有限公司];AUW220D十万分之一分析天平(日本岛津)。
1.2 试药
大卫颗粒(陕西兴邦药业有限公司,批号分别为:160902、161001、161101,规格:6 g/袋);绿原酸对照品(批号:110753-201415,纯度96.2%)、黄芩苷对照品(批号:110715-201619,纯度93.5%)、连翘酯苷A对照品(批号:111810-201606,纯度93.4%)、连翘苷对照品(批号:110821-201615,纯度94.9%)、甘草苷对照品(批号:111610-201607,纯度93.1%)、金银花对照药材(批号:121060-201608)、连翘对照药材(批号:120908-201216)、柴胡对照药材(批号:120992-201509)均购自中国食品药品检定研究院;硅胶G薄层板、硅胶H薄层板(青岛海洋化工厂);乙腈为色谱纯;水为屈臣氏纯净水;正丁醇、甲醇、三氯甲烷等其他试剂均为分析纯。
2 方法和结果
2.1 定性鉴别
2.1.1 连翘 供试品溶液:取本品4 g,加60 mL水溶解,用40 mL水饱和的正丁醇振摇提取,重复操作3次,将正丁醇液合并,分取60 mL供柴胡鉴别用,剩余的正丁醇液减压回收至干,残渣加2 mL甲醇溶解,即得[8]。
对照药材溶液:取连翘对照药材0.5 g,加40 mL水,加热微沸1 h,滤过,滤液按供试品溶液制备方法制备,即得。
对照品溶液:取连翘苷对照品10 mg,加甲醇制成0.5 mg·mL-1的溶液,即得。
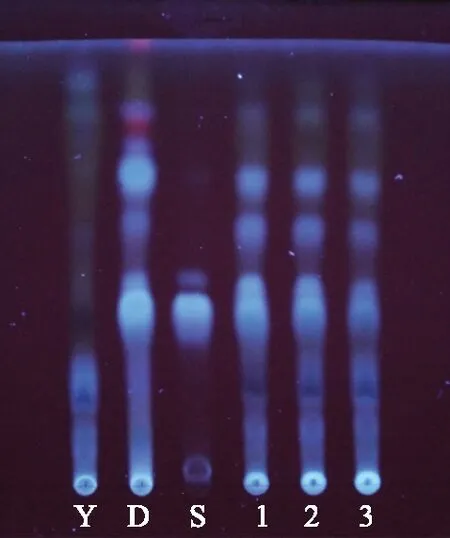
阴性对照溶液:按大卫颗粒处方取缺连翘的其他药味,按其制剂工艺制备缺连翘的阴性样品,并按供试品溶液制备方法制备,即得。吸取上述4种溶液各5 μL,分别点于同一硅胶G薄层板上,展开剂为三氯甲烷-甲醇按体积比5∶1配置,将薄层板展开,取出,晾干,采用10%硫酸乙醇溶液为显色剂,在105 ℃加热至斑点显色清晰。结果阴性对照对检测无干扰,供试品色谱中,在与对照药材、对照品色谱相应位置上,显相同颜色的斑点,见图1。

注:1~3.供试品;D.对照药材;S.对照品;Y.阴性对照。图1 连翘的薄层色谱图
2.1.2 金银花 供试品溶液:取2 g本品,加15 mL甲醇,超声处理20 min,用甲醇补足减失的溶剂量,滤过,取续滤液,即得。
对照药材溶液:取0.5 g金银花对照药材,加15 mL甲醇,按供试品溶液制备方法制成,即得。
对照品溶液:取绿原酸对照品10 mg,加甲醇制成质量浓度为1 mg·mL-1的溶液,即得。
阴性对照溶液:按大卫颗粒处方取缺金银花的其他药味,按其制剂工艺制备缺金银花的阴性样品,并按供试品溶液制备方法制备,即得。
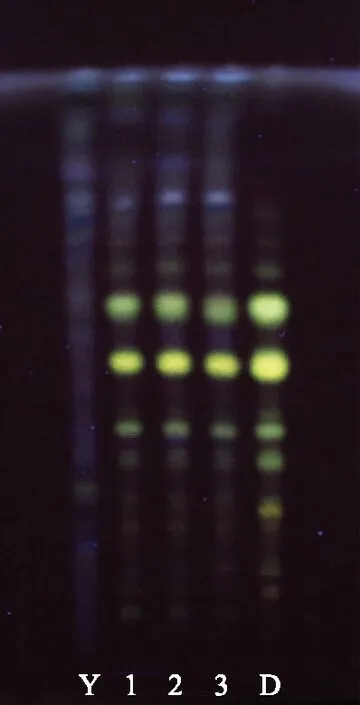
吸取上述4种溶液各6 μL,分别点于同一硅胶H薄层板上,以乙酸丁酯-甲酸-水按体积比7∶2.5∶2.5配置,静置,取上层溶液为展开剂,将薄层板展开,取出,晾干,置365 nm紫外光灯下检视[9]。结果阴性对照对检测无干扰,供试品色谱中,在与对照药材、对照品色谱相应的位置上,均显蓝色荧光斑点,见图2。
注:1~3.供试品;D.对照药材;S.对照品;Y.阴性对照。图2 金银花的薄层色谱图
2.1.3 柴胡 供试品溶液:取2.1.1中剩余的正丁醇液,加入等体积的氨试液洗涤,再用等体积正丁醇饱和的水洗涤,弃去水层,正丁醇液减压回收至干,残渣加1 mL甲醇溶解,即得。
对照药材溶液:取柴胡对照药材0.5 g,加40 mL水,加热微沸1 h,滤过,滤液按供试品溶液制备方法制备,即得。
阴性对照溶液:按大卫颗粒处方取缺柴胡的其他药味,按其制剂工艺制备缺柴胡的阴性样品,并按供试品溶液制备方法制备,即得。吸取上述3种溶液各2 μL,分别点于同一硅胶G薄层板上,展开剂为三氯甲烷-甲醇-水按体积比13∶7∶2配置后,在10 ℃以下放置的下层溶液,将薄层板展开,取出,晾干,采用1%对二甲氨基苯甲醛的硫酸乙醇溶液(1→10)为显色剂,在70 ℃加热至斑点显色清晰后30 min,置紫外光灯(365 nm)下检视[10],结果阴性对照对检测无干扰,供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点,见图3。
2.1.4 甘草 对照品溶液:取甘草苷对照品10 mg,加甲醇制成质量浓度为0.5 mg·mL-1的溶液,即得。
阴性对照溶液:按大卫颗粒处方取缺甘草的其他药味,按其制剂工艺制备缺甘草的阴性样品,并按供试品溶液制备方法制备,即得。
吸取2.1.1项下的供试品溶液及上述两种溶液各2 μL,分别点于同一硅胶G薄层板上,展开剂以乙酸乙酯-甲酸-冰醋酸-水按体积比15∶1∶1∶2配置,将薄层板展开,取出,晾干,置紫外光灯(365 nm)下检视[11]。结果阴性对照对检测无干扰,供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的荧光斑点,见图4。
注:1~3.供试品;D.对照药材;Y.阴性对照。图3 柴胡的薄层色谱图
2.2 含量测定
2.2.1 色谱条件与系统适应性 色谱柱Agilent Eclipse XDB-C18(250 mm×4.6 mm,5 μm);流动相:乙腈(A)-0.2%磷酸水溶液(B),梯度洗脱(0~15 min,13%A,87%B;15~20 min,13%~15%A,87%~85%B;20~28 min,15%A,85%B;28 min~38 min,15%~35%A,85%~65%B;3~50 min,35%A,65%B)[12];流速为1.0 mL·min-1;检测波长为320 nm;柱温35 ℃;进样量为10 μL。在上述色谱条件下,待测成分分离良好,分离度>1.5。
2.2.2 对照品溶液制备 取绿原酸13.70 mg、连翘酯苷A 14.80 mg、黄芩苷17.04 mg,精密称量,置50 mL容量瓶中,加50%甲醇适量使溶解,再加50%甲醇至刻度,摇匀,即得(临用新制)。
2.2.3 制备供试品溶液 取本品适量,研细,取约0.2 g,精密称定,置100 mL容量瓶中,加50%甲醇适量,超声处理20 min,放冷,再加50%甲醇至刻度,摇匀,经0.45 μm微孔滤膜滤过,取续滤液,即得。
2.2.4 专属性试验 按大卫颗粒处方取缺金银花、连翘、黄芩的其他药味,按其制剂工艺制备阴性样品,并按2.2.3供试品溶液制备方法制备,制成阴性对照溶液。分别取对照品溶液、供试品溶液、阴性对照溶液各适量,按2.2.1项下色谱条件进样测定,记录色谱图,见图5。结果,供试品中绿原酸、连翘酯苷A、黄芩苷对照品在相同保留时间处有相同色谱峰,并与其他组分峰基线分离较好,且相应的阴性对照不出峰。表明该方法可以用于该制剂中3种成分的测定。

注:A.对照品;B.供试品;C.阴性对照;1.绿原酸;2.连翘酯苷A;3.黄芩苷。图5 大卫颗粒的高效液相色谱图
2.2.5 线性关系考察 分别精密吸取2.2.2项下对照品溶液0.2、0.4、0.8、1.0、1.5、2.0 mL,分别置于10 mL容量瓶中,加50%甲醇定容制成系列对照品溶液。在2.2.1项下色谱条件进样测定,记录峰面积,以峰面积(Y)为纵坐标,以待测成分浓度(X)为横坐标进行线性回归,绘制标准曲线,得绿原酸回归方程:Y=31 859.2X-30 585,r=0.999 8;连翘酯苷A回归方程:Y=777 875X-3316,r=0.999 9;黄芩苷回归方程:Y=36 127.9X+839.84,r=0.999 9;结果表明,绿原酸在5.228~52.28 μg·mL-1,连翘酯苷A在5.92~59.20 μg·mL-1,黄芩苷在6.816~68.16 μg·mL-1与峰面积呈良好的线性关系。
2.2.6 进样精密度 取2.2.3项下供试品溶液(批号:160901),按2.2.1项下色谱条件连续进样测定6次,记录峰面积并计算绿原酸、连翘酯苷A、黄芩苷含量。结果3种成分的平均质量分数分别为2.9、5.0、7.2 mg·g-1,峰面积RSD分别为1.10%、0.76%、1.28%,表明仪器精密度良好。
2.2.7 重复性试验 精密称取同一批样品(批号:160901)适量,共6份,按2.2.3项下方法制备供试品溶液,再按2.2.1项下色谱条件进样测定,记录峰面积并计算绿原酸、连翘酯苷A、黄芩苷含量。结果3种成分的平均质量分数分别为2.9、4.9、7.2 mg·g-1,峰面积RSD分别为1.62%、1.83%、1.38%,表明本方法重复性良好。
2.2.8 稳定性试验 取2.2.3项下供试品溶液(批号:160901),分别于制备后 0、2、4、8、12、24 h进样测定,记录峰面积并计算绿原酸、连翘酯苷A、黄芩苷含量。结果3种成分的平均质量分数分别为2.8、4.9、7.2 mg·g-1,峰面积RSD分别为0.96%、1.25%、0.84%,表明供试品溶液室温放置24 h稳定。
2.2.9 加样回收率试验 精密称取同一批样品(批号:160901)适量,共6份,分别加入实际质量浓度为105.84 μg·mL-1绿原酸对照品溶液2 mL,276.46 μg·mL-1连翘酯苷A对照品溶液2 mL,254.37 μg·mL-1黄芩苷对照品溶液3 mL,按2.2.3项下方法制备供试品溶液,再按2.2.1项下色谱条件进样测定,记录峰面积并计算加样回收率,结果见表1。
2.2.10 含量测定 取3批本品各适量,分别按2.2.3项下方法制备供试品溶液,再按2.2.1项下色谱条件进样测定,记录峰面积并计算绿原酸、连翘酯苷A、黄芩苷含量,结果见表2。

表1 加样回收率试验结果(n=6)

表2 样品含量测定结果(n=3) mg·g-1
3 讨论
在TCL鉴别中,原标准中仅收载了绿原酸的薄层鉴别方法,本研究在此基础上,对处方中6味药材的TCL鉴别方法均进行了研究,其中黄芩薄层鉴别中,采用对照药材作对照,阴性样品有干扰,采用黄芩苷对照品作为对照无干扰,但考虑到含量测定中对制剂中黄芩苷的含量进行测定,故本研究未收载黄芩的薄层鉴别,另外紫苏叶的薄层鉴别中阴性样品对鉴别有干扰,未能建立较好的TCL鉴别方法。综上,通过研究最终建立了大卫颗粒中连翘、金银花、柴胡、甘草的TLC鉴别方法。同时,为了考察TLC系统的耐用性,实验中对不同温度(4、25、35 ℃)、不同厂家薄层板进行实验。结果表明,TLC图斑点清晰,分离度好,阴性对照均无干扰。
根据中药质量标准的研究要求,复方中药含量测定方法中,应首选君臣药、贵重药、毒性药,不建议选取含量较低成分。连翘、金银花为大卫颗粒的君药,绿原酸为金银花中的主要功效成分,可作为大卫颗粒的质量控制性成分,连翘中的主要成分有连翘苷、连翘酯苷等,现有大部分连翘制剂多测定其连翘苷含量,但大量研究表明,连翘酯苷是连翘发挥抗菌、抗病毒等作用的主要功能成分[13],且连翘中连翘酯苷的含量远高于连翘苷[14-15],所以本研究将连翘酯苷A也作为大卫颗粒的质量控制性成分之一。另外,黄芩药材中指标性成分黄芩苷含量较高(≥9.0%),且黄芩苷常作为黄芩相关制剂的含量测定成分[16]。综上,本研究选择绿原酸、连翘酯苷A、黄芩苷作为大卫颗粒的质量控制性成分,并采用高效液相法对3种成分同时进行含量测定。
含量测定方法耐用性研究中,我们考察了不同厂家、不同品牌的3种色谱柱:Agilent Eclipse XDB-C18(250 mm×4.6 mm,5 μm)、Phenomenex Luna 5u C18(2)(250 mm×4.6 mm,5 μm)、Ultimate XB-C18(250 mm×4.6 mm,5 μm)对制剂中绿原酸、连翘酯苷A、黄芩苷成分的分离情况,3种色谱柱对测定成分的分离均较好;另外实验中还考察了流动相的不同酸度(甲醇-0.1%、0.2%、0.3%磷酸水、乙腈-0.2%、0.5%、0.8%醋酸水溶液)、不同柱温(30、35、40 ℃)对分离效果的影响,结果表明,流动相pH值与柱温在一定的范围内波动,对成分的测定无影响,说明该方法耐用性良好。
综上,本研究所建立的标准可用于大卫颗粒的质量控制,为其质量标准提高提供参考。
