含双金属碳化物复合材料的制备及其析氢性能研究
2019-05-13
(浙江工业大学 化学工程学院,浙江 杭州 310014)
氢能源作为一种清洁、高效的绿色能源在替代传统化石燃料上被寄予厚望。目前制氢的方法主要有生物制氢、化石燃料制氢、光催化分解制氢和电解水制氢等[1]。其中电解水制氢技术具有绿色环保、产品纯度高、原料来源丰富便捷和生产工艺简单等优点,但其对电量需求大,而电催化剂的使用可以明显降低析氢过电位,从而有效地改善这一缺点[2]。目前最高效的析氢催化剂是铂系贵金属,这类催化剂在析氢反应中起始电位接近于零,过电位低且反应速率较快[3-5],但其储量稀少且市价昂贵,因此开发高效、稳定的能代替铂系贵金属的新型析氢催化剂既迫切又重要。在各类析氢催化剂中,过渡金属碳化物如碳化钼、碳化钨等,因具有丰富的来源、低廉的成本、良好的热稳定性、优异的酸碱稳定性、类铂的表面电子结构和催化性能[6-7],从而吸引了广大研究者的注意,目前这方面的研究也取得了一些重大突破[8-12]。随着研究的深入,发现在过渡金属碳化物中引入其他的过渡金属元素,利用不同的过渡金属间的协同效应,能优化氢吸附的吉布斯自由能和M—H键能[13-15],且这类材料中的界面结构也能加速水裂解和活泼氢结合的过程,这些因素都有助于提升析氢电催化性能[16]。此外,碳材料如无定形碳、石墨烯、碳纳米管和碳布等,作为一种被广泛应用的载体,具有良好的导电性能、较大的比表面积和易调控的多孔结构,有利于均匀分散催化剂活性组分和加快传质过程,对催化性能的提高具有很好的促进作用[11-12,17-18]。鉴于此,科学家们花费了大量的时间和精力致力于合成多元过渡金属碳化物类复合材料,如Bukola等[14]制备的Co,N掺杂的碳化钨双功能催化剂在HER和ORR体系中均有良好的催化性能,这主要得益于Co的引入能够减小催化剂动力学进程的阻碍。Wang等[19]设计了一种新型的Ni/Mo合金催化剂 (MoxC-Ni@N-Carbon),该催化剂中金属Ni的添加不仅有助于介孔结构的形成,而且提高了介孔碳的石墨化程度,还促进了高活性的γ-MoC物相的形成,使该催化剂表现出了优异的析氢性能。这些成果表明过渡金属碳化物类复合材料在析氢催化方面具有巨大的潜力。目前这方面研究的一个巨大挑战是这类材料的制备过程复杂、反应原料众多,且通常需要高温环境和较多步骤才能完成,严重限制了其进一步的发展和推广。
鉴于此,笔者选用含过渡金属类的杂钨酸Na10[Co4(H2O)2(PW9O34)2]·27H2O (以下简写为{Co4P2W18O70})与有机胺十六烷基三甲基溴化铵 (CTAB) 作为反应原料,希望通过有机胺提供碳源和弱还原气氛,通过含过渡金属的杂钨酸提供多种金属来源的同时还发挥过渡金属对于碳的石墨化的催化作用[15],从而达到简单高效地制备多元过渡金属复合材料的目的。结果表明:采用上述策略,在较低温度下煅烧即可制备多元过渡金属复合材料。XRD和电镜测试表明该复合材料的主要物相组成为WOx,Co6W6C和无定型碳载体,且产物的形貌、微观结构和催化性能能够通过煅烧温度、煅烧时间等因素得到有效的调节。此外笔者对产物的析氢催化性能也做了初步研究。上述合成策略简单易行并具备较强的普适性,对制备其他类似的过渡金属复合材料具有很好的借鉴作用。
1 实 验
1.1 试剂与仪器
钨酸钠 (Na2WO4·2H2O, AR),磷酸氢二钠 (Na2HPO4, AR),硝酸钴 (Co(NO3)2·6H2O, AR),氯化钠 (NaCl, AR),十六烷基三甲基溴化铵(C19H42NBr, AR),无水乙醇(C2H5OH , AR),乙醚(C4H10O, AR),浓硫酸(H2SO4,AR),氢氧化钾(KOH, AR),氮气(Ar, 纯度99.999%),Nafion乳液 (Nafion, 5%,质量分数), 异丙醇(C3H7OH, AR)。
管式电阻炉,上海实验电炉厂;电化学工作站,CHI 660E,上海辰华仪器公司。
1.2 Na10[Co4(H2O)2(PW9O34)2]·27H2O晶体的合成
该多酸化合物采用Yin等[20]使用的合成方法,具体过程如下:将10 g钨酸钠、0.48 g磷酸氢二钠、1.95 g硝酸钴溶于50 mL去离子水中,搅拌均匀后用37%的盐酸调节该溶液的pH值,当调节pH至7后再继续搅拌15 min;将得到的深紫色溶液回流2 h后,称取15 g氯化钠趁热溶解在该溶液中,过滤后将滤液静置24 h得到紫色晶体。将晶体收集后再进行重结晶,静置24 h后得到多酸晶体{Co4P2W18O70}。
1.3 WO2/Co6W6C@C的制备
将0.54 g,0.1 mmol {Co4P2W18O70}溶于30 mL去离子水中配成溶液A,另外将0.73 g,2 mmol CTAB溶于30 mL去离子水中配成溶液B,将溶液B缓慢滴加至溶液A至浅紫色沉淀不再产生为止,继续将混合液搅拌1 h后分别用去离子水、乙醇、乙醚洗涤离心并干燥,得到浅紫色粉末。将上述浅紫色粉末平铺于瓷舟内,将瓷舟放置于管式炉内,在氮气气氛中进行程序升温煅烧。具体的升温程序如下:首先从室温升温到220 ℃,在该温度下保温1 h,接着再从220 ℃升温至410 ℃,在该温度下保温1 h,随后继续升温到800 ℃并保温1 h或3 h,待煅烧程序结束后,将管式炉在氮气气氛下自然冷却到室温,取出瓷舟并收集瓷舟内黑色粉末即为最终产物。为了探索煅烧温度和保温时间对最终产物的影响,在对比试验中分别选取了700 ℃和750 ℃为最终煅烧阶段,保温时间为1 h或3 h条件下制取的一系列样品。
1.4 表征及测试
采用粉末X射线衍射 (XRD) 分析材料的物相组成,X射线衍射仪为荷兰PANalytical 公司生产的X’Pert PRO型,X射线源为Cu靶Kα辐射 (λ=0.154 1 nm),管电压40 mV,管电流40 mA,扫描范围为10°~80°,扫描速度2.4 (°)/min。采用Hitachi S-4700 II型扫描电子显微镜 (SEM) 研究样品的微观形貌,辐射源为Cu Kα,操作电压为15 kV。采用英国雷尼绍公司的inVia型拉曼光谱仪 (Raman) 对样品内部分子的能级结构信息进行表征,测试过程采用波长为532 nm的激发光,测试范围为1 000~3 000 cm-1,检测时间为20 s。
析氢电催化性能测试采用上海辰华CHI 660E电化学工作站,所有电化学测试体系均为三电极体系,其中工作电极为覆有催化剂的玻碳电极(直径d=3 mm),辅助电极为碳棒,酸性和碱性环境中的参比电极分别为氯化银电极 (Ag/AgCl) 和氧化汞电极 (Hg/HgO),酸性电解液为0.5 mol/L的H2SO4溶液,碱性电解液为1 mol/L的KOH溶液。工作电极的制备方法如下:将3 mg催化剂粉末分散在90 μL异丙醇和10 μL Nafion (质量分数为5%)中并超声形成均匀的悬浮液,取2 μL该悬浮液滴加在预处理过的玻碳电极表面,待自然风干后即可作为工作电极使用,催化剂的负载量为0.857 mg/cm2。本文中电化学数据中的电势均为相对于可逆标准氢电极的电势 (vs.RHE),其转化公式为
酸性电解液:E(RHE)=E(Ag/AgCl)+(0.222 4+0.059 pH)
碱性电解液:E(RHE)=E(Hg/HgO)+(0.098+0.059 pH)
在进行电化学测试前,预先通入氮气30 min且整个电化学测试均在氮气气氛中进行,此外所有数据均未经过iR校调。
2 结果与讨论
2.1 样品的XRD分析
图1显示的是在不同煅烧温度和不同煅烧时间下得到的样品的XRD图。从图1(a)中可以看出:当煅烧温度为800 ℃时,所制得样品的主要物相为WO2(JCPDS#01-032-1393)和Co6W6C (JCPDS#00-023-0939)。其中衍射角2θ位于25.788°,36.775°和52.939°处的特征衍射峰分别归属于WO2(JCPDS#01-032-1393)的 (011),(200)和(220) 晶面。衍射角2θ位于23.083°,32.902°,36.041°,40.606°,43.167°和47.254°处特征衍射峰则分别归属于Co6W6C (JCPDS#00-023-0939)的 (220),(400),(331),(422),(511)和(440)晶面。值得注意的是,在800 ℃下将保温时间由1 h增加到3 h时,最后所得样品的XRD图谱中还出现了较弱的WC (JCPDS#00-025-1047) 的特征衍射峰,其衍射角2θ位于31.475°,35.627°和48.267°,分别对应于WC (JCPDS#00-025-1047) 的 (001),(100) 和 (101) 晶面。
观察由对比试验所制备的样品的XRD图谱 (图1b),可以发现:当煅烧温度为700 ℃,保温时间为1 h时,所制得的样品的主要物相组成为WO3,其衍射角2θ位于23.637°,33.64°,41.464°,48.431°,54.572°和60.25°处的特征峰分别是由WO3(JCPDS#00-046-1096)的 (200),(220),(222),(400),(420) 和 (422)晶面的衍射引起的。当煅烧温度为700 ℃,保温时间为3 h时,样品中开始有少量的CoWO4(JCPDS#00-015-0867) 出现。此外,当煅烧温度升高到750 ℃时,所制得样品的XRD图谱中不仅出现了明显的CoWO4(JCPDS#00-015-0867) 特征衍射峰,还出现了少量WO2(JCPDS#01-032-1393) 特征衍射峰,说明煅烧温度的升高和保温时间的延长对于物相组成的改变起主要作用。上述实验结果还说明:随着煅烧温度由700 ℃升高到800 ℃,样品中发生的物相转变主要为WO3+CoWO4→W+Co→WO2+Co6W6C,这可能是由于有机胺CTAB在温度升高后分解速率加快,分解产物中可能含有大量的有机小分子化合物,从而形成一种碳化还原氛围,WO3被还原为WO2的同时,还使得CoWO4在渗碳作用的影响下转变为Co6W6C[21-22]。
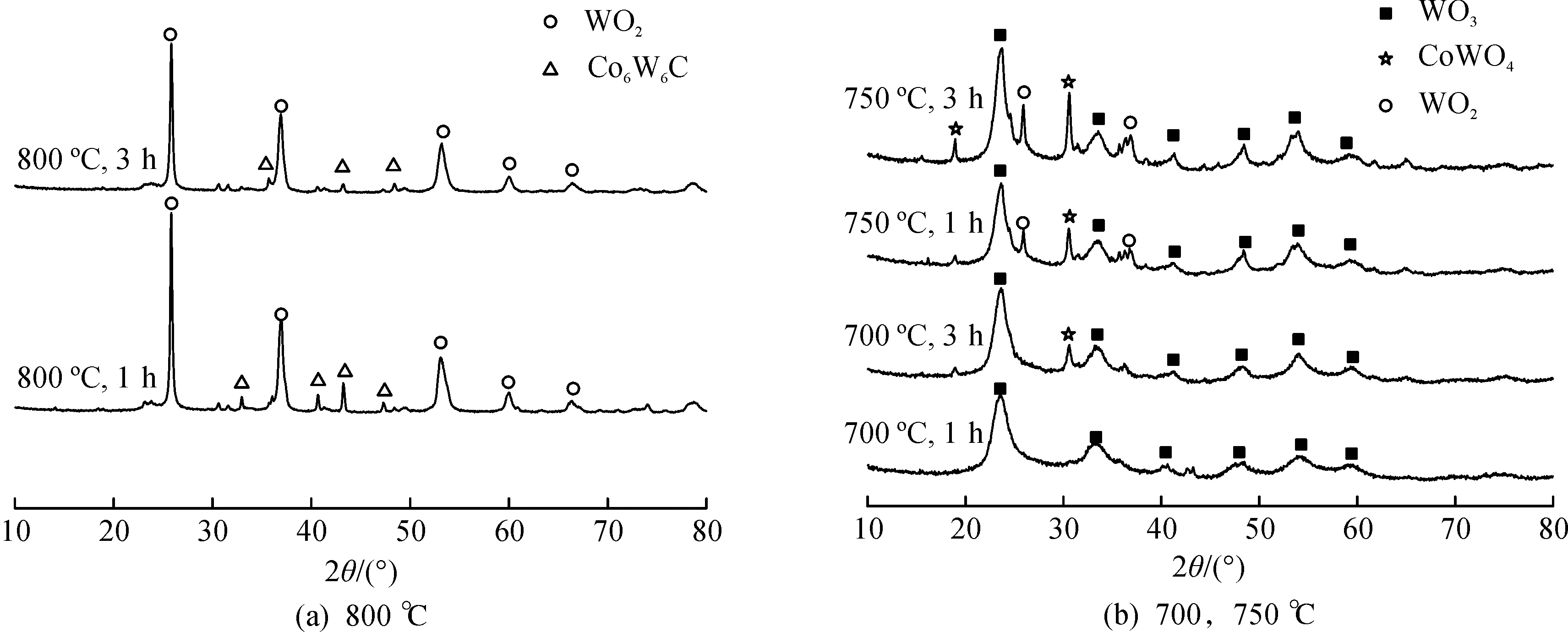
图1 不同煅烧温度和煅烧时间下得到的样品XRD谱图Fig.1 XRD patterns of samples calcined at different temperatures for different time
2.2 样品的SEM形貌表征
从图2可以看出:在800 ℃,1 h的煅烧条件下 (图2e) 制得的样品主要为团聚纳米颗粒,其尺寸大约为30~50 nm,另有少部分大块体存在。改变煅烧条件为800 ℃,3 h (图2f),所得样品的形貌变成类海胆状的聚集纳米棒,这些纳米棒长度在400 nm、直径在50 nm左右。在对比试验中,700 ℃ ,1 h (图2a)、700 ℃,3 h (图2b) 和750 ℃,1 h (图2c) 条件下制得的样品的形貌均为致密堆积的体块状,并没有特定的形貌。而750 ℃,3 h (图2d) 条件下制得的样品则在堆积的大块上开始出现尺寸为20~30 nm的团聚颗粒。这些实验现象说明:较高的煅烧温度和较长的保温时间有利于形成具有特定形貌的样品。结合相关的文献报道和前述XRD结果推测,这可能是由过渡金属元素Co对碳的石墨化催化作用造成的。具体说来,当温度较低时,过渡金属元素Co主要以CoWO4的形式存在,体系中没有任何单原子状态的Co,因而无法催化石墨化碳的形成;而当温度升高时,CoWO4在分解形成Co6W6C的过程中,单原子态的Co能够催化形成石墨化的碳原子,进而赋予最终产物某种特定的形貌。上述Co对碳的石墨化催化作用也与笔者选用含有过渡金属的杂钨酸以及含有大量C的有机胺作为反应原料的初衷相符合。

图2 不同煅烧温度和煅烧时间下得到的样品的SEM图Fig.2 SEM images of samples calcined at different temperatures for different time
2.3 样品的拉曼光谱表征
从图3可以看出:拉曼光谱图中都有两个主要的特征峰(G峰和D峰),其中G峰(1 586 cm-1)是由sp2杂化碳的伸缩振动所产生的,可以反映材料的对称性和结晶完整程度,而D峰(1 354 cm-1)是碳材料的缺陷峰,是由碳材料的无序度引起的[23]。从图中可以看出:所有煅烧条件下的样品均在1 360~1 600 cm-1出现了明显的D峰和G峰,表明杂钨酸有机无机杂化前驱体经煅烧后所制得的样品均含有无定型碳。
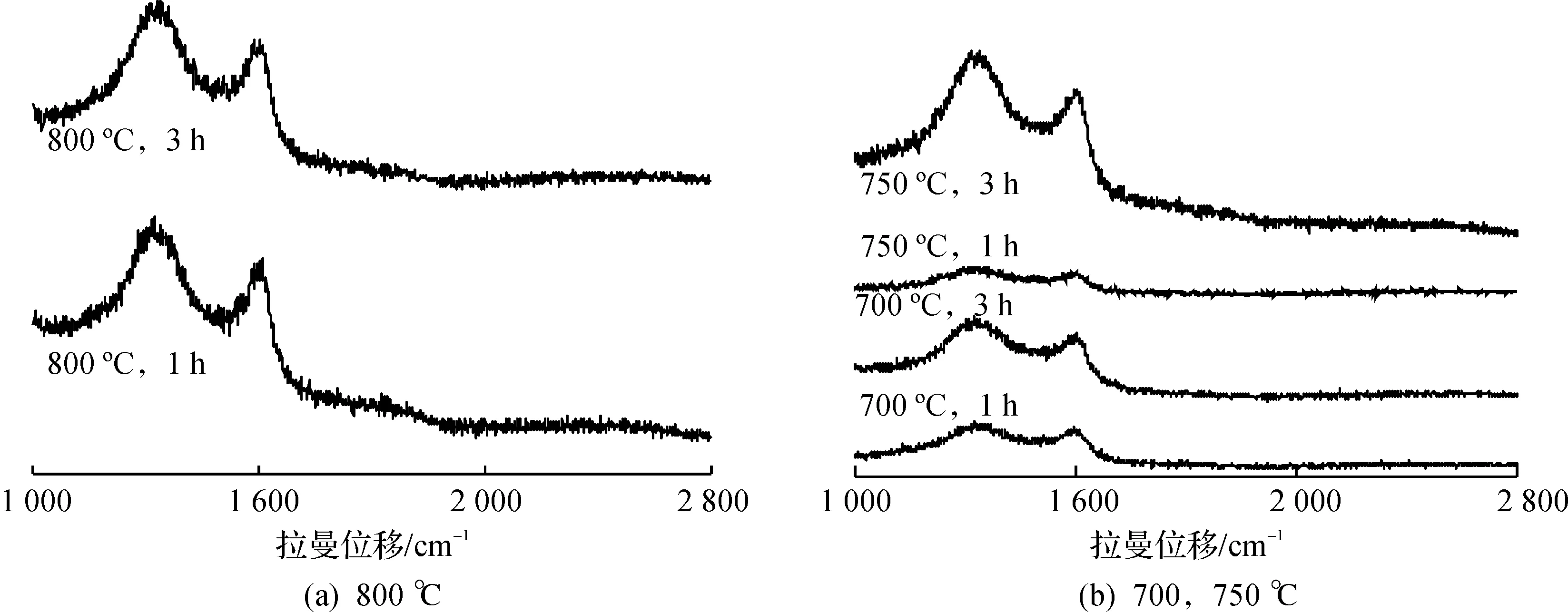
图3 不同煅烧温度和煅烧时间下得到的样品的拉曼谱图Fig.3 Raman patterns of samples calcined at different temperatures for different time
2.4 电化学测试与分析
2.4.1 酸性体系下各样品的析氢活性评估
为了评估所制备的样品的电催化性能,笔者测试了样品在不同电解液中的电化学产氢活性。图4(a)显示的是采用不同样品制备的工作电极在25 ℃,0.5 mol/L的H2SO4溶液中的线性扫描伏安曲线(LSV),扫描速度为5 mV/s。从图中可以看出:800 ℃,1 h和800 ℃,3 h的煅烧条件下制备的WO2/Co6W6C@C的析氢性能一般,起始电位分别为340 mV和360 mV,在电流密度为10 mA/cm2时的过电位均为540 mV,塔菲尔斜率分别为119 mV/dec和146 mV/dec,说明其析氢反应主要采取的是Volmer-Heyrovsky[24]机制。上述结果表明:800 ℃,1 h条件下获得的样品的析氢电催化活性要优于800 ℃,3 h条件下获得的样品,可能的原因在于800 ℃,3 h的样品中有部分Co6W6C转化成了WC。由于Co6W6C中含有两种金属元素,相对于仅含有一种金属元素的WC,Co6W6C中Co与W两者之间的协同效应使其能发挥出更好的电催化活性,这也与文献报道中的结果一致[14]。而在对比试验中所制备的样品则表现出了较差的析氢活性,普遍都具有相当高的起始电位和过电位 (表1)。结合XRD和SEM分析测试结果推测,造成这种差别的原因可能在样品的物相组成和微观形貌上。对比试验中所获得的样品主要物相为WO3@C,几乎没有或者只有很少的具有催化活性的物相Co6W6C存在,此外这些样品主要为大块状的聚集体,非常不利于催化活性位点的暴露和传质过程的进行,因此其析氢性能较差。

图4 不同煅烧条件下制得样品的线性扫描曲线图和Tafel图Fig.4 Linear sweep voltammetry and Tafel curves of samples obtained under different calcination conditions
表1 不同煅烧条件下制得的样品在酸性体系中的电催化析氢活性
Table 1 Samples’ HER electrocatalytic performances in 0.5 mol/L H2SO4
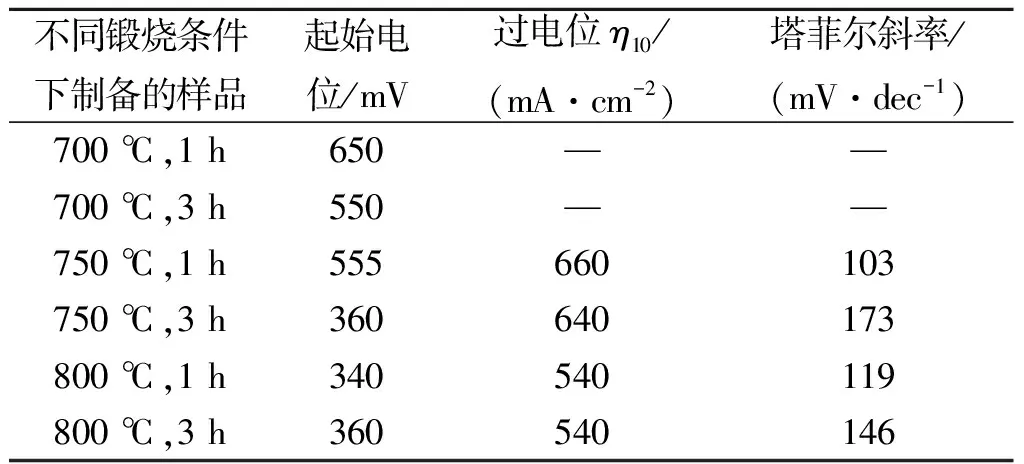
不同锻烧条件下制备的样品起始电位/mV过电位η10/(mA·cm-2)塔菲尔斜率/(mV·dec-1)700 ℃,1 h650——700 ℃,3 h550——750 ℃,1 h555660103750 ℃,3 h360640173800 ℃,1 h340540119800 ℃,3 h360540146
2.4.2 碱性体系下各样品的析氢活性评估
图5(a)显示的是采用不同样品制备的工作电极在25 ℃,1 mol/L的KOH溶液中的线性扫描伏安曲线(LSV),扫描速度为5 mV/s。从图中可以看出:800 ℃,1 h和800 ℃,3 h的煅烧条件下制备的WO2/Co6W6C@C的析氢性能较好,起始电位分别为375 mV和305 mV,在电流密度为10 mA/cm2时的过电位分别是475 mV和450 mV,塔菲尔斜率分别为65 mV/dec和132 mV/dec,说明析氢反应主要是Volmer-Heyrovsky机制。和酸性体系中的析氢性能不同,800 ℃,3 h条件下获得的样品在碱性体系中的析氢电催化活性要稍高于在800 ℃,1 h条件下获得的产品的析氢活性,可能的原因在于800 ℃,3 h的样品中有部分Co6W6C转化成了WC。虽然Co6W6C的电催化析氢活性较WC的高,但后者比前者在碱性环境中的稳定性更好,因此WC总体表现出了略优的析氢活性。相比其他煅烧条件下得到的产品,煅烧条件为800 ℃,1 h时得到的产品塔菲尔斜率较低,主要是由于产品含有较多的双金属碳化物Co6W6C,使得反应中增长同样的电流密度时所需要的过电位较小。而对比试验中所制备的样品的析氢活性则一般,普遍都具有较高的起始电位和过电位 (表2)。这与其在酸性体系中的析氢表现相一致。值得注意的是,所有的样品在碱性体系中的析氢性能皆优于其在酸性体系中的析氢性能,这对碱性条件下析氢催化剂的开发比较有吸引力。
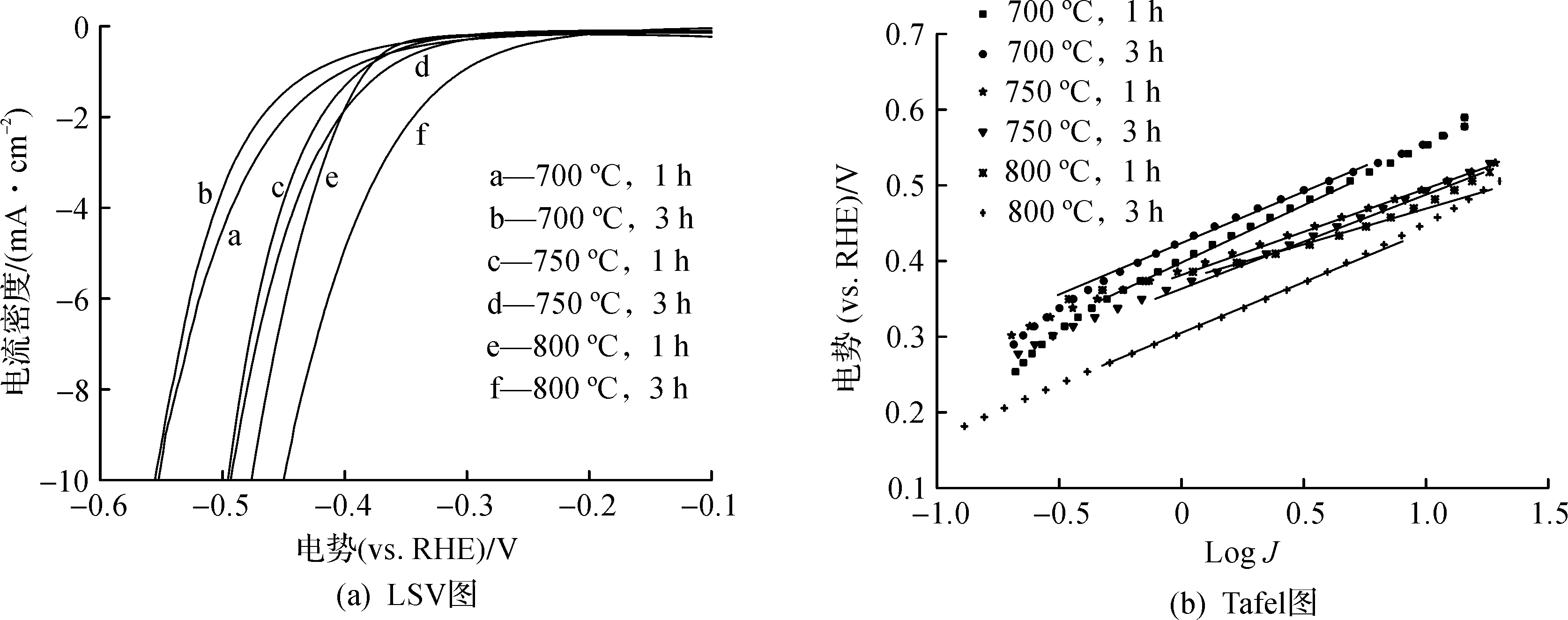
图5 不同煅烧条件下制得样品的线性扫描曲线图和Tafel图Fig.5 Linear sweep voltammetry and Tafel curves of samples obtained under different calcination conditions
表2 不同煅烧条件下制得的样品在碱性体系中的电催化析氢活性
Table 2 Samples’ HER electrocatalytic performances in 1 mol/L KOH
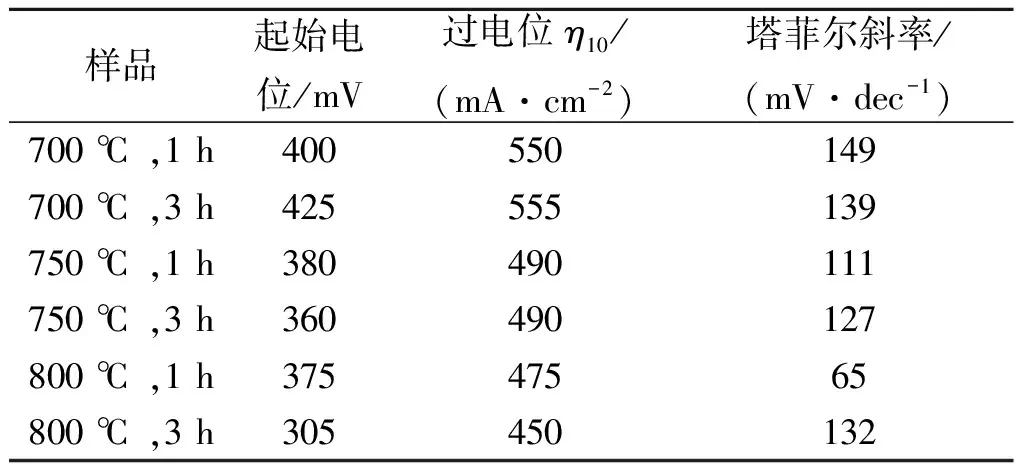
样品起始电位/mV过电位η10/(mA·cm-2)塔菲尔斜率/(mV·dec-1)700 ℃ ,1 h400550149700 ℃ ,3 h425555139750 ℃ ,1 h380490111750 ℃ ,3 h360490127800 ℃ ,1 h37547565800 ℃ ,3 h305450132
3 结 论
以含有过渡金属的杂钨酸({Co4P2W18O70})和CTAB作为反应原料,采用简单的煅烧法设计性地制备了含有双金属碳化物Co6W6C的复合材料。初步的电催化析氢性能表明:所得材料在酸性和碱性体系中均有较好的催化活性,且其在碱性条件中的析氢性能更好。笔者的合成策略主要是通过CTAB来提供丰富的碳源和其在高温分解时形成弱还原气氛,通过杂钨酸提供原子级别上预混合的双金属来源,从而达到在较低的温度下即可制备含有双金属碳化物的目的。这一策略简单易行,对于合成其他类似的多元金属复合材料具有很好的借鉴意义。
