CRISPR/Cas9系统中引导RNA的研究进展
2019-05-09李江耿立召许建平
李江 耿立召 许建平
(先正达生物科技(中国)有限公司,北京 102206)
基因编辑是目前生命科学研究的一个热点,CRISPR/Cas9(The clustered regularly interspaced palindromic repeats/CRISPR-associated 9)是基因编辑技术中的一个重要工具。目前广泛使用的CRISPR/Cas9是细菌和古细菌的对抗外源DNA入侵的一种防御系统,可将入侵的噬菌体基因组DNA等外源核酸序列切除[1]。自2013年首次报道CRISPR/Cas9在生物体内实现基因编辑以来[2-3],由于其操作简单,效率高,所以该系统广泛应用于动植物,成为药物开发、疾病治疗和农作物品质改良等领域的一个重要基因编辑工具,具有广泛应用前景[4]。
基因编辑中所用的CRISPR/Cas9系统属细菌II型的CRISPR/Cas9,由Cas9蛋白和引导RNA(Guide RNA,gRNA)两种分子组成,其中Cas9蛋白是一种依赖于引导RNA分子的核酸切割酶,只有装载引导RNA后才能激活自身识别和切割基因组DNA的功能。而引导RNA除激活Cas9活性外,还含有一段与靶基因组DNA反向互补的20 个核苷酸序列,将Cas9蛋白/引导RNA复合体定靶向位于目的DNA序列[1]。目前关于CRISPR/Cas9系统的研究综述多集中于工作机理、Cas9蛋白的结构与工作模式,以及CRISPR/Cas9系统的应用发展,而对CRISPR/Cas9中的引导RNA研究进展还缺乏系统性回顾。因此,本文将从引导RNA的结构,产生方式以及对基因编辑频率的影响这几个方面,对CRISPR/Cas9系统中的引导RNA研究进展进行综述。
1 引导RNA的序列与结构
在细菌中,CRISPR/Cas9的引导RNA由两条RNA分子组成,CRISPR RNA(CrRNA)和trans activating CRISPR RNA(TracrRNA)( 图 1-a)。crRNA的5′端包含20个碱基与噬菌体基因组DNA互补,并由重复间隔序列串联构成,转录成一条长的RNA分 子(Precursor crRNAs,pre-crRNAs) 后, 经 过RNA 酶III加工过程产生一系列短的40 nt左右的包含间隔序列的成熟crRNA。tracrRNA 具有与crRNA互补的一段序列,crRNA与tracrRNA结合形成部分双链互补的两个RNA分子复合体[1]。Cas9蛋白首次在体外证明具有切割功能时发现其切割活性是由两条引导RNA分子参与产生的,只加入CrRNA不 能使Cas9蛋白实现对质粒DNA的切割,只有再加入TracrRNA时才能使Cas9体外切割质粒DNA,获得与CRISPR/Cas9体内相同的生物学活性[5]。CrRNA 与TracrRNA 有两个RNA分子组成,crRNA 5′端20个碱基与靶基因互补,与crRNA配对结合后促进CrRNA的成熟。虽然CrRNA与TracrRNA通过互补序列形成的双链分子与Cas9结合后产生的基因编辑在原核生物中应用较好,但在真核生物中因技术复杂难以进行应用。为了便于操作,科学家将CrRNA的3′端与TracrRNA的5′端通过GAAA的核苷酸序列相连,并缩短了双链互补结合区域仅保留4个核苷酸对,形成48 nt的单引导RNA分子(图1-b),并通过延长3′端37个碱基后(图1-c),获得编辑效率很高的单引导RNA分子结构。目前,85nt的单引导RNA序列结构是CRISPR/Cas9在基因编辑中广泛使用的一个分子结构[2-3,5-8]。
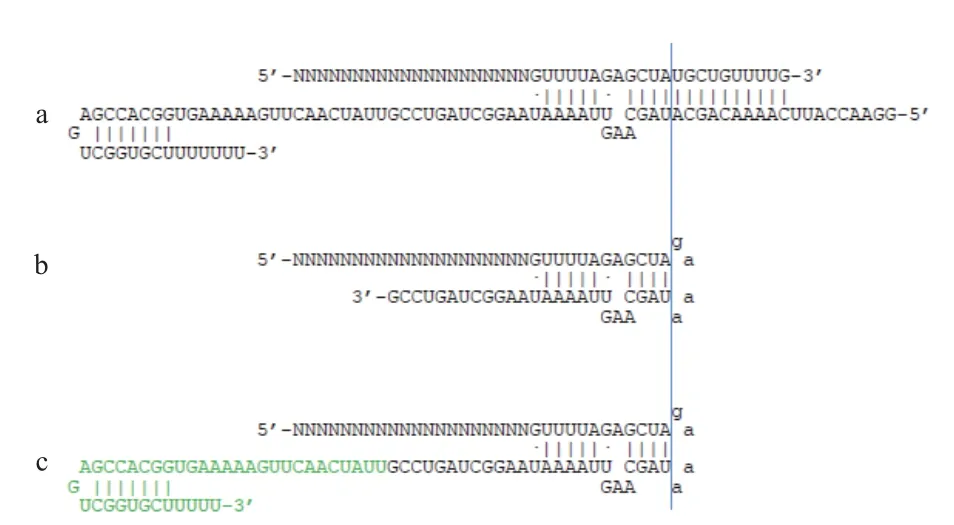
图1 引导RNA的序列结构变化[9]
目前关于引导RNA的结构研究以是以单引导RNA序列为模型获得。引导RNA的序列可分为两部分,5′端1-20个碱基是与目标DNA互补的Protospacer序 列, 它 与 PAM(Protospacer adjacent motif)序列共同决定Cas9蛋白目的DNA序列上的定位,在PAM上有的3-4位核苷酸之间产生双链断裂。Protospacer之后的核苷酸为引导RNA的折叠结构(Scaffold fold),含有多个RNA二级结构。在这些RNA二级结构中,双链互补区域的14个碱基由CrRNA与TracrRNA的重复序列反向互补形成,其中G27、G28、A41、A42、G43和U44不配对而产生一个突起(Bulge),突起上游的双链序列称为下位茎(Lower stem),下游的双链序列成为上位茎(Upper stem)。上位茎下游含有3个茎环组成的结构,依次为 Nexus,Hairpin1 和 Hairpin2[10-11]。对引导RNA的二级结构在不同文章中的命名不完全相同,但对应的结构是一致的。通过对Cas9蛋白结合引导RNA和目标DNA序列的形成的晶体结构解析发现,引导的5′端序列与目标DNA序列形成的复合体被Cas9蛋白包裹于充满负电荷的多肽内部,引导RNA的第10-20个核苷酸称为种子序列(Seed region),与模板DNA互补形成有序而牢固的双链结构,当种子序列存在2个及以上碱基的与目的DNA序列产生错配时,Cas9蛋白不能与目的DNA序列牢固结合无法对DNA序列产生切割[12]。引导RNA中的反向互补的双链重复序列结构被Cas9蛋白的识别结构域(Recognition,REC)和核酸酶结构域(Nuclease,NUC)以序列依赖的方式识别。此结构中的非互补突起部分及相邻的核苷酸是Cas9蛋白识别的核心序列,而末尾的C30∶G39和A32∶U37不被Cas9蛋白识别而突出于Cas9蛋白表面,提示此部分双链互补序列是经RNA酶III加工产生的。引导RNA双链互补结构下游的三个茎环结构是稳定Cas9蛋白与引导RNA复合体的重要序列。其中Nexus的序列中有部分核苷酸(52、53和59-61)是被Cas9蛋白的REC结构域和NUC结构域中的1103-1107 位的氨基酸结合,被PAM激活结构域(PAM-interacting,PI)所识别,与双链重复互补序列中的突起结构共同参与激活Cas9蛋白对PAM序列的识别,引导RNA打开Cas9蛋白的结构去识别PAM序列[13]。其余两个茎环结构Hairpin1和Hairpin2的大部分序列暴露于Cas9蛋白表面,只有Hairpin1中的63-65、67和Hairpin2中的92位核苷酸被Cas9蛋白的NUC结构域识别[10],这两个茎环可以提高Cas9蛋白与引导RNA复合体的稳定性,并且这段区域中的核苷酸能容忍较大范围的改变,是改造引导RNA的一个可行区域[11-12,14]。可见,引导RNA的双链复合体和Nexus是Cas9蛋白行使功能所必需的结构,而Hairpin1和Hairpin2以及之间的5个连接碱基可帮助稳定Cas9蛋白与引导RNA及目标DAN形成的复合体,突变将影响Cas9蛋白的切割效率。
引导RNA的序列特征在Type II的 CRISPR-Cas9系统中具有保守性。在Streptococcus和Lactobacillus中分离到的41 种Cas9基因中,尽管Cas9的蛋白序列根据一致性可分为3组,但引导RNA中CrRNA与TracrRNA反向重复序列结合的双链序列高度保守,都存在不配对的核苷酸突起结构以及相邻的上下位茎序列,是II型CRISPR/Cas9特有的结构。但核苷酸组成和上下位茎的长度在不同来源的Cas9中差异很大,其中较短的下位双链序列长度保守,较长的上位双链序列长度在不同种类的菌中变异较大。但最为保守的部分为TracrRNA的第一个茎环,甚至在不同菌中都含有高度一致的碱基,如A52和C55与Cas9蛋白的1 103-1 107 位的氨基酸结合[10]。对S. thermophiles中的两个同源Cas9蛋白Sth1Cas9和Sth3Cas9以及对应的引导RNA序列CRISPR1 sgRNA和CRISPR3 sgRNA进行置换,未能检测到Sth1Cas9和Sth3Cas9的切割活性。然而当CRISPR1 sgRNA含有 CRISPR3 的Protospacer 序列后可以使Sth3 Cas9产生切割活性。进一步对两种引导RNA的茎环同时进行互换后发现,人工产生的引导RNA不能使原对应的Cas9蛋白产生切割活性,但使异源的Cas9产生了切割活性。而只互换其中的一个茎环不足以产生这种效果[10]。说明引导RNA的序列结构在不同类型的CRISPR/Cas9中具有独特性,表明CRISPR/Cas9系统进化具有不同的分支;也表明引导RNA对CRISPR/Cas9系统发挥功能具有非常重要的决定作用。
2 引导RNA的产生方式
引导RNA在细菌和古细菌中由体内的RNA 转录系统产生,但在CRISPR/Cas9应用于基因编辑技术时,需要在体内表达人工设计的引导RNA,引导RNA的5′端1-20个核苷酸对应目的DNA序列不同而做改变,使Cas9蛋白在特定预期位点产生切割。引导RNA的产生是CRISPR/Cas9基因编辑技术中的重要过程,引导RNA需要满足以下要求:(1)引导RNA保持在细胞核内。(2)产生的引导RNA的5′端不能有与目的序列不配对的多个额外核苷酸。针对不同的基因编辑需求,CRISPR/Cas9的引导RNA有不同的产生方式,会对最终基因的编辑效果产生显著影响。
2.1 单引导RNA转录
CRISPR/Cas9人工构建的引导RNA的5′端1-20核苷酸与靶DNA序列互补,不能有额外的多个核苷酸序列,因此引导RNA最初都是由聚合酶III型启动子(Pol III promoter)转录产生。在植物过表达技术中应用广泛的II 型启动子是一类表达强,在基因过表达中广泛使用的一类生物体内源启动子,如CaMV 35S 启动子、玉米泛素启动子(Ubiquitin)启动子等。但这类启动子转录的RNA是前体结构,如mRNA 前体,microRNA前体和一些小的核RNA前体等,这些非成熟的RNA前体会经过体内加工系统,实现5′ 加帽和3′加尾,并且切除内含子。因此II型启动子不能用于CRISPR/Cas9系统中引导RNA的转录[15]。III型启动子是转录5s RNA,tRNA和小的非编码RNA的启动子。在CRISPR/Cas9中常用的Pol III启动子有U3和U6两类,在哺乳动物细胞中产生引导RNA为U6,在植物中产生引导RNA为U3和U6,U3和U6启动子在转录引导RNA产生基因编辑的频率上没有差别[2-3,6,16]。U3 启动子的转录产物第一个碱基固定是A,而U6启动子的转录产物第一个碱基固定是G,因此引导RNA的5′端序列需根据U3或U6的使用进行调整,如5′GN(19)NGG 和 5′AN(19)NGG[17]。在使用 Pol III启动子转录产生引导RNA时,通常一个U3或U6启动子只能产生一个引导RNA分子去实现一个靶位点的切割。当需要产生多个引导RNA时,需要将这类启动子重复使用去驱动多个引导RNA产生[18]。
U3和U6启动子在CRISPR/Cas9基因编辑中广泛用于引导RNA的转录,但具有一定的局限性。U3和U6启动子是组成型表达启动子,不具备组织特异性表达,这导致引导RNA的产生无法在时空上进行调节,不能实现条件诱导性的基因编辑。生物体内的U3和U6启动子分布广、种类多,不同种属之间序列差异大,同一物种中的启动子活性也不同。启动子的有些调控元件位于转录起始位点下游内,容易导致克隆的启动子序列不完整,影响启动子活性[15]。近来也有研究报道通过RNA聚合酶II型启动子转录一条包含Cas9 和引导RNA的转录本,这条转录本中的引导RNA可被RNaseIII途径加工后,与Cas9结合在水稻中产生高频率的基因编辑活性[19-20]。
2.2 多引导RNA转录
当 CRISPR/Cas9要对体内多个靶点实现基因编辑时,需要产生多个引导RNA与目的序列位点结合。多个引导RNA的产生最早是用U3或U6启动子交替重复使用,每个启动子对自身下游的引导RNA序列进行转录。采用此策略在水稻和拟南芥中可同时分别转录6个引导RNA产生,获得16%的纯和突变体的靶位点编辑频率[21]。虽然这个方法可以得到多个靶位点的突变体,但利用U3/U6重复表达多个引导RNA有以下不足:(1)由于引导RNA长度很短,并且要求引导RNA的5′末端为A/G,3′末端需要为5个及以上的poly(T)作为终止信号,克隆构建策略有限,无论是采用DNA合成还是多个片段拼接,引导RNA转录单元串联的载体十分具有挑战性。目前文章报道采用的策略均在Golden Gate的技术上做改进,最多可将5个引导RNA表达单元装入一个载体,而6个以上的引导RNA表达单元组装效率很低[16]。(2)串联重复的引导RNA表达单元包含启动子后在300-600个碱基左右,其中只有与目的基因序列互补的20 个碱基作为Protospacer有变化,其余序列均为重复序列。过多的重复序列容易造成载体在细菌和农杆菌中的不稳定。(3)由于多引导RNA转录结构的串联,容易在植物体内诱发基因沉默,造成引导RNA的低水平或不表达。以上这些因素使U3或U6启动子重复使用产生多个引导RNA的基因编辑技术存在很大的挑战[21]。
近年来,多个引导RNA的产生是CRISPR/Cas9基因编辑技术中的一个研究热点。在生物体内存在可以从一个RNA转录本中产生多个RNA分子的机制,如多顺反子mRNA前体在转录后加工过程中被RNA酶剪切后可产生多个独立的引导RNA分子。因此,可以利用生物体内的RNA剪切加工过程从一个RNA转录本中同时产生多个引导RNA分子。目前报道在CRISPR/Cas9系统中利用3种RNA剪切过程实现多个引导RNA的产生:来源于Pseudomonas aeruginosa的Csy4的RNA切割酶,tRNA序列介导的内源RNA酶剪切和病毒来源的核酶剪切系统。Csy4作为外源RNA切割酶,多个引导RNA被其20个核苷酸的识别序列间隔开,Csy4识别这段序列并切割间隔序列的3′末端,可释放两个Csy4识别序列之间的引导RNA分子,产生的引导RNA的5′端没有额外核苷酸存在而3′端带有Csy4的20nt的识别序列[22]。2017年,有研究将Csy4通过2A肽与Cas9蛋白融合表达,引导RNA通过Csy4识别序列串联,可在体内通过能从一个RNA转录本上同时产生12个引导RNA分子[23]。生物体内还存在一类tRNA加工的RNA 剪切加工系统,常用的是tRNAGly是一段77nt 的核苷酸形成的一个包含3个茎环结构的一段RNA序列,其5′端含有一个RnaseP的识别切割位点,3′端含有一个RnazeZ的识别切割位点,在体内通过内源核酸酶将tRNA序列的两个位点切割从而释放引导RNA。当多个引导RNA通过在5′和3′端连接有77 nt 的tRNA序列进行串联时,核酸酶加工切割过程可从一个RNA分子上释放多个引导RNA,产生的引导RNA的3′端经RNaseZ切割后残留6个tRNA序列的核苷酸而5′不含额外的核苷酸[24-25]。tRNAGly序列在动植物中均存在,介导的多个引导RNA转录释放技术在CRISPR/Cas9技术中得到了广泛使用,通过此方法实现了同时对动植物体内多个位点进行基因编辑,有报道最多可一次产生8个引导RNA分子[23]。此外,与tRNAGly序列释放引导RNA的过程类似,在引导RNA的5′端和3′端各加上一种核酶序列,5′端是Hammerhead(HH)type ribozyme,3′端 是 Hepatitis delta virus(HDV)ribozyme,这种结构形成的引导RNA分子称为 RGR(Ribozyme-gRNA-Ribozyme,RGR)。RGR可利用核酶序列间隔,将多个引导RNA分子串联转录后,引导RNA两侧的核酶序列被体内核酸酶识别并切除,从而释放有活性的引导RNA分子,但这两种核酶的切割活性较tRNA结构要低很多[23],并且在需要引入动物病毒的核酶序列,在植物应用中有很大局限。但tRNA结构不同,这两种核酶在SP6启动子介导的体外转录过程中,可被SP6 RNA聚合酶在转录过程中识别并切除[26]。
以上3种策略中,Csy4和tRNA介导的多个引导RNA在体内产生并实现多靶位点的基因编辑频率相当,而远远高于HH ribozyme和HDV ribozyme核酶产生的引导RNA。有报道表明Csy4核酸酶不会对植物体产生负表型影响,而且Csy4的识别序列只有20个核苷酸,远短于tRNA的77个核苷酸序列,有利于载体的构建和稳定[23]。tRNA的切割加工是生物体的一个基本活性过程,因此tRNA介导的多个引导RNA的产生可广泛用于动植的基因编辑,是多位点CRISPR/Cas9基因编辑的一个研究热点[15,23-25]。而 HH ribozyme 和 HDV ribozyme核酶能被SP6识别并切割,因此更适用于体外转录产生多个引导 RNA 的应用[15,26]。
2.3 体外产生引导RNA
在CRISPR/Cas9基因编辑技术中,Cas9和引导RNA可以在体外产生后组装成蛋白核酸复合体(Ribonucleoproteins,RNPs),导入细胞体内实现靶位点DNA编辑。体外产生引导RNA主要由用商品化的T7体外转录试剂完成,引导RNA的DNA序列5′端含有T7启动子序列和转录起始位点[27-28]。体外转录的引导RNA可以是单分子形式,也可以是CrRNA和TracrRNA两个分子。引导RNA也由化学合成的方法在体外产生,常用的方法是固相基质上如利用 2′-silyl,2′-bis-methylther等化学合成方法。但单引导RNA的分子长度接近100个核苷酸,合成的成本和难度大,因此通常采用CrRNA和TracrRNA两个分子的形式[29]。
3 优化引导RNA提高基因编辑效率
CRISPR/Cas9虽然广泛应用于各种动植物体的基因编辑,但在一些生物体中仍存在效率低的问题,如小麦等[30]。此外,同源重组介导的核苷酸定点插入和替换的效率依赖于Cas9的切割效率。群体中足够多的双链断裂(Double strand break,DSB)是实现同源重组的必要条件,而引导 RNA 5′端的序列结构是影响同源重组效率的另一因素[31]。因此,提高CRISPR/Cas9的基因编辑效率对这项技术的广泛和深入应用有重要意义。引导RNA作为CRISPR/Cas9的重要组成之一,引导RNA的序列、高级结构以及表达方式对CRISPR/Cas9的基因编辑效率有显著影响[32]。
3.1 引导RNA序列组成对基因编辑效率的影响
引导RNA的序列由与目标DNA序列互补的Protospacer和Scaffold 两部分组成。5′端的1-20个核苷酸是与目标序列互补的一段序列,其中的核苷酸组成会影响基因编辑效率,当G和C出现频率高而A出现频率低,尤其是GC含量高于50%时,可以提高引导RNA序列与靶基因序列位点结合的稳定性,提高Cas9的切割效率;尤其是靠近靶序列的PAM位点的核苷酸中,20位核苷酸偏好G而避免C,19位核苷酸避免C时,可显著提高引导RNA产生的编辑效率。此外,引导RNA通常由RNA聚合酶III型的U3或U6启动子转录产生,RNA序列中连续4个及以上的尿嘧啶将成为这类启动子的终止信号,可导致转录的提前终止[33]。在引导RNA内部位于Lower stem的第23-26位4个连续尿嘧啶序列UUUU[23,26-30]是U3或U6启动子潜在的终止信号,当这四个尿嘧啶分别被突变为A,C,G时,均可提高CRISPR/Cas9的基因编辑效率,尤其是突变为C和G时较A的基因编辑效率高很多;并且第26位的尿嘧啶突变为C时,提高的编辑效率较其他3位的突变最为明显[9]。这种优化的引导RNA序列在水稻基因编辑中得到了应用[14]。
3.2 引导RNA序列结构对基因编辑效率的影响
引导RNA的二级结构是被Cas9蛋白识别并产生功能的重要序列。利用荧光探针淬灭技术研究不同序列缺失的引导RNA与Cas9蛋白体外结合时发现,缺失第一个茎环(Nexus)将导致Cas9蛋白不能结合引导RNA,这与Cas9蛋白结合引导RNA的晶体结构相吻合,缺失第2个茎环(Hairpin1)和第3个茎环(Hairpin2)时,Cas9蛋白结合引导RNA的效率要降低很多,尤其是与总RNA共同混合时尤其明显。说明引导RNA的二级结构是CRISPR/Cas9产生基因编辑的重要部分,还对引导RNA在细胞内特异性被Cas9蛋白识别结合有作用[34]。应用于CRISPR/Cas9基因编辑系统中人工创造的引导RNA序列中,双链互补区的序列比天然crRNA:tracrRNA的双链互补区截短了10个碱基对,目前关于引导RNA二级结构优化提高CRISRP/Cas9基因编辑效率的研究集中在这部分序列。当延长这部分序列从1、3、5、8和10个碱基对时,发现延长5个碱基对时引导RNA的基因编辑效率可达到最大[9,35]。在水稻中,延长双链结合区5个碱基对并叠加第26位的尿嘧啶突变为C时,这种类型的引导RNA结构比现有序列的基因编辑提高13倍[14]。此外,对引导RNA的3′端添加G3U3或G2U1的特定核苷酸序列,通过提高引导RNA在体内的稳定性而提高CRISPR/Cas9的切割效率并降低脱靶率[36]。
3.3 RNA聚合酶II启动子转录引导RNA对基因编辑效率的影响
在CRISPR/Cas9基因编辑中,引导RNA通常由RNA聚合酶III型转录产生,但由于这类启动子的强度较常用的II型启动子强度低,限制了CRISPR/Cas9的编辑效率。在多引导RNA产生中发展来的几种多顺反子RNA切割系统,可以使引导RNA的5′端不受RNA聚合酶III启动子限制,而是用强度更大的RNA聚合酶II型启动子转录引导RNA。在番茄原生质体中,用CmYLCV(Cestrum Yellow Leaf Curling Virus promoter)启动子驱动tRNAGly和Csy4介导的两种引导RNA转录,对黄色荧光蛋白(Yellow fluorescent protein,YFP)的编辑效率比U6启动子驱动的引导RNA要高2倍[21]。考虑到RNA聚合酶II型启动子的35S 和Ubiquitin启动子常用于驱动Cas9表达盒,为了避免载体含有重复的大片段,目前有多个RNA聚合酶II型启动子以供引导RNA转录,除前文提到的植物病毒来源的CmYLCV启动子,还有细菌来源的M24和Nos以及植物来源的 AtUbi10,OsAct1,PvUbi1和 PvUbi2等, 甚 至有组织特异性的启动子如Arabidopsis Ec1.2 和YAO promoter可供引导RAN实现植物卵细胞,囊胚和花粉中的特异表达[21,37]。
4 总结与展望
CRISPR/Cas9系统作为基因编辑重要的一个工具,已经广泛用于各种生物体的特定核苷酸的缺失和改变,以及特定基因的转录调控等。引导RNA作为CRISPR/Cas9系统中的核心元件之一,对其序列和结构的研究不仅加深人们认识CRISPR/Cas9的自然分类、工作原理,而且为CRISPR/Cas9系统的优化和拓展具有重要价值[38-39]。最近报道在引导RNA的3′端添加用于同源重组介导的RNA形式的供体序列,在Cas9蛋白产生的双链断裂缺口处完成供体RNA链与目的DNA序列链的置换,可以大幅提高核苷酸的定点编辑的效率[40]。此外,引导RNA的保守序列部分为新CRISPR-Cas系统的发掘提供重要的信息和证据[41]。
