监管科学的起源
2019-05-05杨悦
文/杨悦
一、背景
科学与政策之间的相互作用有着悠久的传统。在19世纪末和20世纪初,科学界在美国社会决策过程中发挥的作用是微不足道的。政府当局负责制定规则,同时宗教领袖也将宗教解释为规则;彼时,科学要么不被考虑,要么在决策过程中扮演次要角色。另一个关键原因是在那个时期,科学要么不够先进,要么与信仰(包括迷信)混在一起,因此很难涵盖社会主体的利益。随着各行业的发展,公众逐渐萌生了对相关业务的监管愿望,包括采矿、制造业、农业、空气污染、饮用水、水污染和食品安全等。立法者、监管者和公众都认识到,规范这些活动需要获得相关的科学信息[1]。
1970年12月尼克松总统将联邦机构的不同组织联合起来,成立了美国环境保护署(EPA)。然而EPA成立之初,一些法律授权到期或不再适用,法律法规亟待不断修订,监管机构意识到,适用于本机构的法规制定依据尚不充分,各机构必须根据尚不完整的科学信息做出决策。“监管科学”一词在此期间被创造出来,旨在满足监管机构的科学需求。
二、“监管科学”术语的由来
第一个认识到监管科学性质的研究者是Alvin Weinberg,他在1970年首先用监管科学的理念评估电离辐射的影响[2]。他在1972年发表的《Science and Trans-Science》一文中指出,一些问题需要用科学方法解决,但科学方法又无法完全回答,为此他提出“跨科学/转化科学(Trans-Science)”的概念。尽管从认识论的角度讲,它们是关于事实的问题,可以用科学语言描述,却无法用科学来回答;它们超越了科学的界限。这是监管科学理念的原始萌芽[3]。
“监管科学(Regulatory Science)”一词的真正出现在1970年12月EPA成立后不久,该术语首先出现在一份未标注日期的内部备忘录中,由Alan Moghissi博士提出,用于描述该机构制定法规所用的科学。最初人们不接受这个术语,理由是在制定法规时使用科学并不罕见。有人认为“科学就是科学”,无论其应用如何。1985年春,Alan Moghissi在弗吉尼亚州成立了一个非营利性的监管科学研究所,其目标是“在科学与监管体系之间进行科学研究[4]。监管机构的建立并未导致人们认识到需要新的科学学科[1]。Alan Moghissi继续对监管科学进行广泛应用的阐述,这一理念得到了FDA的认同。
监管科学的要旨不是得到真理(truth)本身,而是实现“可用的真理(serviceable truth)”。(The point for regulatory science is not to get at the truth perse.The goal is to achieve a serviceable truth.)
将监管科学进行学科构建的是美国哈佛大学从事科学技术研究 的Sheila Jasanoff教授。她在1990年首次对“regulatory science”这个词进行了广泛而深入的描述,试图找到一个能与现有“研究科学”(research science)进行区分的标志,她认为这是一门包含了科学、社会和政治相互关系的学科,而不仅仅是监管机构和其他一些决策制定者从诸多独立、客观和科学的研究中去发现、挖掘并引导而形成的学科。监管科学的要旨不是得到真理(truth)本身,而是实现“可用的真理(serviceable truth)”。(The point for regulatory science is not to get at the truth perse.The goal is to achieve a serviceable truth.)美国FDA于1991年使用监管科学这一概念解决医药等“科学产品”(包括有型产品、知识和信息),因此使监管科学得到了 FDA 的重视,并将其确定为 FDA 在 21 世纪重点推动的学科[2]。
另一位对监管科学有贡献的专家是日本的内山美树(Mitsuru Uchiyama)博士。1987年,曾在日本国立卫生研究院(National Institutes of Health Sciences)工作的内山博士被认为是日本第一个提出监管科学概念的人。在那之后,内山博士用日语写了许多关于监管科学的短文,在1996年,他在一本国际期刊上用英文发表了一篇关于监管科学的分析性评论文章,提出监管科学是根据人类健康目标来优化科学技术的科学。内山博士认为,在美国,监管科学是为回答政治问题而产生的科学,而在日本,监管科学主要被用来讨论医药科技研发[5]。
三、“监管科学”含义界定
关于“监管科学”,不同组织有不同的含义界定,笔者将其梳理如下,并附英文原文。
FDA科学委员会将监管科学描述成公众健康(public health)机构为履行职责所需的基于科学的决策过程[6]。
The FDA Science Board's 2007 Science and Mission at Risk report describes regulatory science as a science-based decisionmaking process needed to fulfill the responsibilities of a public health agency.
FDA前 局 长Margaret Hamburg2009年指出监管科学用来评估和评价产品安全性的科学和工具。
“…the science and tools we use to assess and evaluate a product's safety.” Margaret Hamburg, FDA Commissioner
美国监管科学研究所所长Alan Moghissi 2009年 在《The Scientist》发文称,监管科学是科学在各个层面上对社会决策过程的独特应用。
“Regulatory science is a unique application of science, at all levels, to the societal decision process.” Alan Moghissi, President, Institute for Regulatory Science
2010年,FDA在《NIHFDA合作加速快速通道产品创新的声明》(《Announcement of NIH–FDA Collaboration to Fast-Track Innovations to the Public)》中将监管科学描述为研发和使用新工具,标准和方法,以便于更有效地研发产品,更有效地评估产品的安全性、有效性和质量。
The development and use of new tools, standards and approaches to more efficiently develop products and to more effectively evaluate product safety, efficacy and quality.
美国宾夕法尼亚大学医学院Garret FitzGerald 2010年 指 出,监管科学是通过获取和分析足够的数据,以指导与批准安全有效的治疗产品、器械和化妆品以及确保食品供应安全和营养价值相关的决策。
“The acquisition and analysis of data sufficient to inform decision making pertinent to the approval of safe and effective therapeutics, devices and cosmetics and ensuring the safety and nutritional value of the food supply.” Garret FitzGerald, University of Pennsylvania School of Medicine
美国南加州大学药学院对监管科学的描述为:监管科学将生物医药产品研发的监管和法律要求与确保产品安全、有效的科学研究联系起来[7]。
Regulatory Science relates the regulatory and legal requirements of biomedical product development to the scientific research needed to ensure the safety and efficacy of those products.” Academic website for the University of Southern California School of Pharmacy.
美国医学研究所(Institute of Medicine)认为监管科学是应用科学方法来改进新药、生物制品和在需要上市前审批的器械的研发、审查和监督的科学[8]。
Regulatory science is the application of the scientific methods to improve the development, review, and oversight of new drugs, biologics, and devices that require regulatory approval prior to dissemination.
美国国立卫生研究院(National Institutes of Health)认为监管科学促进新的或改良的工具、方法、标准以及更易理解的应用科学的开发、评价和获得,并在产品全生命周期中持续改进产品的安全性、有效性和质量的评价[9]。
Regulatory science fosters the development, evaluation, and availability of new or improved tools, methods, standards, and applied science that support a better understanding and improved evaluation of product safety, quality, effectiveness, and manufacturing throughout the product life cycle.
EMA对将监管科学的定义为:适用于评估医药产品质量、安全性和有效性的一系列科学学科,并在整个药物生命周期内为监管决策提供信息。它包括基本和应用医学及社会科学,并有助于制定监管标准和工具[10]。
Range of scientific disciplines that are applied to the quality, safety and efficacy assessment of medicinal products and that inform regulatory decision-making throughout the lifecycle of a medicine.It encompasses basic and applied medicinal science and social sciences, and contributes to the development of regulatory standards and tools.
目前,FDA将监管科学定义为研发新工具、新标准和新方法,以评估受FDA监管的产品的安全性、有效性、质量和性能的科学[11]。
Regulatory Science is the science of developing new tools, standards, and approaches to assess the safety, efficacy, quality, and performance of all FDAregulated products.
关键路径计划的内容主要是通过全社会的共同努力来发现并优先解决影响医药产品开发的一系列最紧迫的问题,从而使当代生物医学基础研究所取得的重大成果能迅速转化为提高公众健康的新医药产品。
四、关键路径计划
2004年3月,FDA在广泛征询社会各界意见的基础上,发表了名为《创新/停滞:新医药产品关键路径上的挑战与机遇》的白皮书,分析了目前新医药产品研发及审批过程中存在的问题,探讨了产生这些问题的原因并提出了相应的解决方案[12]。在这份白皮书中,FDA正式提出了“关键路径计划”(The Critical Path Initiative,CPI)。关键路径计划的内容主要是通过全社会的共同努力来发现并优先解决影响医药产品开发的一系列最紧迫的问题,从而使当代生物医学基础研究所取得的重大成果能迅速转化为提高公众健康的新医药产品。提出关键路径计划的最主要目的在于通过创造新的、能更准确地判断和预测新医药产品的安全性及有效性的工具,确保最新的生物医学基础研究成果能更快、更确定、更低成本地转化为新的,更有效的治疗手段。
FDA将关键路径定义为新药、新生物制品及新医疗器械的研发过程中的关键性环节。当企业选定某个新化合物、新的生物制品或器械的原型设计进行新产品研发时,就开始进入关键路径了,然后这类新医药产品需要经过严格的临床前及临床试验、医药产品申报、审评、批准后,才能生产上市。在新药的研发过程中,只有很少的一部分能顺利通过关键路径最终上市。
在实施CPI后两年的时间里,FDA通过与企业界、学术界及其他社会团体广泛合作并充分参考各方意见,于2006年3月正式发表了“关键路径机遇清单”(FDA' s Critical Path Opportunities List)[13]。在报告中,FDA总结了自己在CPI推出后两年时间里所进行的工作,列举了一些对改进新医药产品研发和审评过程具有重要意义的领域,并呼吁企业界、学术界及其他社会团体和个人共同合作来解决这些问题;在清单中FDA介绍了76个对新医药产品的研发及审评有重要意义的领域,诸如如何将基因组学、蛋白质组学、影像学及生物信息学等应用到新医药产品的研发过程中来,提高安全性及有效性预测的准确度。关键路径计划中六大优先发展领域为:更好的评价工具(better evaluation tools);简化的临床试验(streamlining clinical trials);生物信息学的应用(harnessing bioinformatics);21世纪的产业化生产开发(moving manufacturing into the 21st century);针对公众健康急需的产品(developing products to address urgent public health needs);特殊的高风险人群用产品(specific atrisk population-pediatrics)。2009年FDA发 布CPI主 要成就报告(Report on Key Achievements)中,展示了FDA在三个重要方向包括生物标记物和其他科学工具、简化临床试验、确保产品安全性等方面取得的成就,以及多个具体领域诸如临床试验转化项目(clinical trials transformation initiative,CTTI)、治疗乳腺癌的新生物标记物等方面取得的进展。
五、监管科学的重要作用和发展途径
在关键路径计划等取得重大成就的基础上,2010年,FDA启动了监管科学计划,将监管科学融入到FDA监管活动的方方面面[14]。
FDA将监管科学定义为:“发展评价监管产品安全性、有效性、质量和功能的新工具、标准和方法的科学”。监管科学的推进和创新是FDA保护和提升公众健康核心任务的基础。作为一个以科学为基础的机构,FDA必须使用最佳的、最可及的科学数据来支持监管决策,以此改善有益于公众健康的FDA监管产品并增强对所有产品的监管。重视监管科学发展的现实意义就是在于促进了药物创新,为预防、治疗和诊断疾病提供了新手段[15]。
2011年8月17日,FDA发布了《促进FDA监管科学:战略计划》(Advancing Regulatory Science at FDA: A Strategic Plan), 在FDA监管药品、食品、化妆品等产品上推行以科学为基础的监管理念[16]。该计划确定了缩小为支持监管决策制定所要求的科学知识之间关键差距的策略,包括提供新的医药产品研究工具、模型;提高临床试验的质量和效率;识别和评估缺乏最佳终点指标试验领域的临床终点和相关生物标记物等手段,解决监管科学和创新的需求,促进产品开发和审评过程。通过缩小上述差距,FDA的监管科学计划可以促进新型科学技术和基础科学的发现向实际诊断、治疗、治愈疾病的方法转化,从而降低了研发产品的时间、复杂性和成本。同样地,通过提供用以审评的创新型工具,监管科学将会帮助FDA审评人员更好地获得新产品的数据并更好地评价新产品。监管科学工具对于加速患者需要的新型、安全且有效的治疗手段的开发是必不可少的。
在该计划中FDA明确了8个重点科学优先领域(Science Priority Areas),包括:①为 了提高产品安全性的毒理学现代化;②通过采取促进临床评价和个体化医疗创新的激励政策,改善产品开发及患者治疗结果;③促进产品生产改进和扶持质量提高的新方法;④确保FDA做好对创新技术评价的准备工作;⑤通过信息科学使用多种数据,提高健康结局水平;⑥实施新的预防为主的食品安全体系,促进患者健康保护;⑦促进保护美国乃至全球健康和安全免受威胁的医疗对策发展;⑧加强社会和行为科学,帮助消费者和专业人员使用产品时做出明智的决策。2013年FDA又新增了第9个重点发展领域,即加强全球产品安全网络。
六、FDA战略计划的提出
1.FDA使命与战略
FDA通过确保人用药品、兽用药品、生物制品、医疗器械、食品、化妆品、放射性制品的安全性、有效性,从而行使其保护和促进公众健康的职责。FDA通过鼓励创新,使药品、食品更加安全、有效、可及,同时通过为大众提供正确的、科学的信息,使他们可以选择让自己更健康的药品和食品。为实现上述使命,FDA制定了2014-2018年的战略重点(strategic priorities)和战略计划(每四年更新),统领着FDA及各大中心和办公室的工作(如图1)。
2.FDA 2014-2018年的5个战略重点领域
战略重点领域是FDA发展的核心理念,指导FDA战略计划的实施和战略目标的实现,包括:①监管科学(regulatory Science);②全球化(globalization);③安全和质量(Safety and Quality);④智慧监管(Smart regulation);⑤管理工作(Stewardship)。这5项战略重点是独立的,但也相互关联不可分割(如图2)。其中监管科学是FDA所有工作的核心重点,其他四个重点与之交叉,共同助力FDA实现效益最大化[17]。
监管科学的推进和创新是FDA保护和提升公众健康核心任务的基础。
监管科学的推进和创新是FDA保护和提升公众健康核心任务的基础。
图2 FDA 2014-2018年的5个战略重点领域
全球化要求FDA运用全球化的思维思考、参与全球工作。实现保护美国公众的目标,这取决于FDA在国外的监管能力。FDA必须与国外的监管部门、行业、地区或国际组织一道,鼓励并促进以科学为基础的标准的实施,保证产品的安全和有效性。智慧监管指FDA通过智慧、合理和以科学为基础的监管,构建最合理的监管制度框架同时减少不必要的负担,在鼓励创新的同时实现保护公众健康的目标。智慧监管要求FDA保持动态化,与时俱进,发现和利用最优的科学技术,从而引导有价值的创新的实现,促进行业的公平竞争,减少产品召回,巩固消费者信心,维持FDA在全球创新的领导地位。
安全和质量包括制造产品的规范;将产品提供给消费者供应链的完整性和保护公众的方法,包括为特定类别产品和产品安全报告系统的实验室样品分析。
管理工作强调FDA要利用有限的预算和资源去实现各项计划的实施。随着一系列法案赋予FDA新的监管职责和权力,FDA会继续优先招聘、发展和雇佣高质量的劳动者,改善职员招聘、补偿、培训、评估和雇佣系统和流程。
FDA将通过分层规划的框架实施这些战略重点。首先,FDA高层将这些战略重点纳入年度预算编制和流程规划中。FDA各大中心和办公室按照项目制定明确计划和关键指标,并及时监控,以反映出战略目标和项目实现的进程。进展情况将结合长期目标与战略,通过年度绩效和雇员绩效计划方案以及项目绩效指标(如国会预算办公室的年度绩效指标、用户费用绩效措施以及FDATRACK措施)进行监管;项目执行情况将通过FDA-TRACK行动由高层领导定期审查(每季度审查一次)。
3.FDA 2014-2018年的核心目标及战略措施
在顶层战略计划的基础上,FDA还提出了2014-2018年的四大核心目标(表1),即加强产品监管;改善和保障FDA监管产品的可及性以使公众健康获益;促进利益相关者知晓FDA监管产品的决策信息;强化组织的优良运作和责任机制。实现上述核心目标的具体战略措施如下:
加强产品监管。包括①增强监管科学的使用,以促进标准的发展、分析和决策;②降低制造、生产和分配上的监管风险;③加强对产品问题的监测与监督;④提高识别产品紧急问题的应对能力。
改善和保障FDA监管产品的可及性以使公众健康获益。具体指①提高监管科学用以评估产品的效率;②提高产品开发过程的效率;③提高审评程序的可预见性(predictability)、一致性、透明性和高效性。
提高监管科学用以评估产品的效率在于提高监管活动的科学性和创新性,解决未满足的医疗和公众健康需求,保护患者和消费者。科学技术的快速发展使FDA在监管产品的开发,评估,生产和使用方面发生了根本变化。例如细胞和基因疗法、纳米技术等新产品促进健康改善的同时,也需要新的方法评估这些产品的安全性和有效性。FDA在推动和促进创新的同时必须根据可获得的最佳的科学数据和最好的工具、方法用以评估监管产品的安全性、有效性、质量、公众健康影响和公众使用。为确保美国的全球创新领导地位,FDA将继续提高监管科学能力和高效率地评估监管产品。在2014-2018年,FDA将重点实施以下战略措施:①加强与科研机构、工业和其他监管团体的合作、培训和信息共享;②推动药品研发工具的开发,促进改善生命质量和用于挽救生命药品的开发并且缩短医药产品研发时间,复杂性和成本;③加强基础设施建设提供高质量、最先进的科学研究;④ 推动生物信息学基础设施现代化在系统生物学、食品安全、基因组学、药物基因组学、预测毒理学、神经系统功能和生物医学影像领域应用最新数据。
近年来FDA在审评新药方面取得了巨大进步,一些重大新药审批的效率和质量处于世界领先,然而一些对于公众健康非常重要的药物(如抗生素)的开发仍严重滞后。这些针对特殊疾病、未满足的医疗需求和特殊人群的医药产品的开发成本往往较大,产品开发者也面临着严峻的挑战。在这种背景下,科学创新活动更需要重视对医药产品开发过程的优化,实现医药产品开发的效率和可预见性,从而开发出安全、有效、可及的医药产品。
科学创新活动更需要重视对医药产品开发过程的优化,实现医药产品开发的效率和可预见性,从而开发出安全、有效、可及的医药产品。
提高药品开发过程效率的战略措施,包括:①促进药品开发和试验中使用的评价方法、工具和模型(如动物模型、生理模型、计算机模拟)的开发;②促进罕见病药品开发;③促进FDA和发起人在医药产品开发过程的沟通;④促进先进技术、方法和相关科学发现(如新发现的临床生物标记物,自适应临床试验设计和基因组学)的应用;⑤促进个体化医疗发展;⑥促进新抗菌药的开发。
提高审评过程的可预见性、一致性、透明性和高效性(predictability, consistency,transparency,and efficiency)是促进创新和提高FDA监管产品可及性、促进公众健康的重要组成部分。FDA认为在当今经济形势下,明确这一监管目标极其重要。及时评价新的人用药品、兽药、生物制品和医疗器械的安全性和有效性是FDA保护和促进公众健康的核心使命。用户付费法案(如PDUFA、GDUFA、BDUFA、ADUFA等)规定申请人支付一定费用支持FDA雇佣额外的审评人员和升级信息系统。与此同时,FDA承诺明确的审批目标并在规定时间内采取措施。这些改变极大地改善了审批过程,使FDA能够在严格保证上市药品的安全性、有效性标准的基础上快速审批药品。同时在审评过程中加强监管人员与申请者的沟通对于审批效率的提高也有重要作用。审批周期长有时往往是因为申请材料不完整、缺失严重或是FDA与申请者需要额外的讨论。如果这些问题能够在审评开始时解决,那么就会避免审评资源的浪费。FDA一直致力于通过增加与发起人的有效沟通,促使审批过程透明化,从而减少审评资源浪费。《处方药使用者付费法案》(2012 PDUFA V)允许新分子实体的新药申请(NME NDAs)和创新生物制品申请(BLAs)采用新的审批模式,关键就在于增加审评过程的交流。FDA一直致力于通过自动化标准IT体系,实现人用药品、生物制品等产品全生命周期的沟通交流、审评以及信息管理。在2014-2018年,FDA将 通 过实施以下战略措施来提高审批程序的可预见性、一致性、透明化以及高效性:①药品申请数据采用电子化提交方式提高审评效率;②实施电子化管理审评程序提高审评效率;③提交数据标准化和完整性提高审评效率;④考虑健康差异和健康产出的监管决策;⑤加强与行业和公众在上市前的审批流程和申请提交的状态方面的沟通。
促进利益相关者知晓FDA监管产品的决策信息。①加强社会和行为科学,帮助患者,消费者和专业人员对产品使用作出最明智的决策;②增加患者和产品供应商获得FDA监管产品风险-效益信息的渠道;③提高向公众提供的产品安全和健康信息的质量。
强化组织的优良运作和责任机制,具体指①招募、发展、保留和战略上管理高水平的工作人员;②提高FDA整体运作的效率;③对基础设施进行投资增强机构的效率和能力。
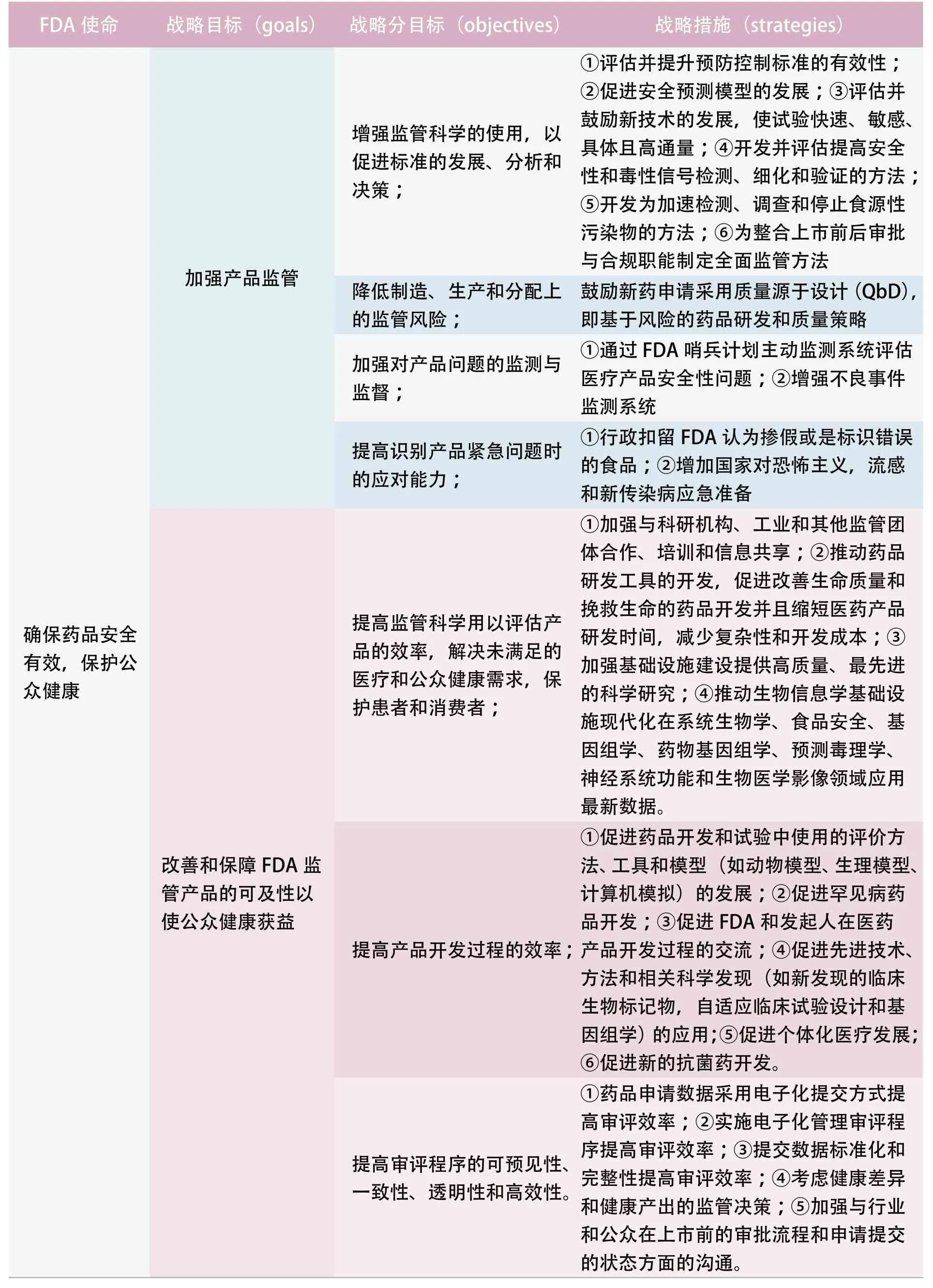
表1 FDA 2014-2018战略重点计划

续表

续表
