基于高通量测序的藜麦连作根际土壤微生物多样性研究
2019-04-29董艳辉于宇凤王亦学聂园军侯丽媛李亚莉王育川曹秋芬吴慎杰秦永军
董艳辉,于宇凤,温 鑫,王亦学,聂园军,侯丽媛,李亚莉,刘 江,任 元,王育川,曹秋芬,吴慎杰,王 斌,秦永军
(1.山西省农业科学院 生物技术研究中心,山西 太原 030031;2.农业部黄土高原作物基因资源与种质创新重点实验室,山西 太原 030031;3.山西省农业科学院 农业资源与经济研究所,山西 太原 030031;4.山西省农业科学院 农作物品种资源研究所,山西 太原 030031;5. 山西省农业科学院 农业环境与资源研究所,山西 太原 030031)
连作是农民常用的一种简单、长期、有效地提高土地利用率获取经济利益的一种做法,藜麦属于苋科植物,作为一种引进于南美的全营养作物,营养价值高,被列为全球十大健康食物之一[1],2008年,我国山西开始引种并试种成功,山西静乐作为藜麦产业的首个生产基地,目前种植面积已经超过0.2万hm2,且甘肃、内蒙古、青海、吉林、河北、宁夏等省份种植面积也在逐渐扩大[2]。由于藜麦作为一种有着较高营养价值的新型杂粮,市场前景比较广阔,相对于其他作物有较高的经济收入,农民对种植藜麦的热情比较高,在土地资源有限的情况下对藜麦进行连年种植,然而,藜麦第2年重茬种植就会出现出苗率低、植株生长缓慢、病虫害加重、产量急剧下降等现象,采取土壤简单消毒、增施底肥等措施后效果也不明显。藜麦连作障碍现象在藜麦大规模种植地区基本都存在,导致个体种植户和藜麦种植公司有着不同程度的损失,科研人员进行了各方面的尝试,但因为对藜麦各方面的研究还处于初级阶段,目前还没有很好的解决办法。马红梅等[3]对连作灵芝土壤微生物研究表明,连作改变了灵芝栽培土壤中原来的微生态平衡,特别是病原真菌改变了原有的土壤微生境,但具体是哪一种菌还有待进行进一步的分子鉴定。薛超等[4]总结了大量前人对连作机制的研究后认为,施用有机肥尤其是将有机肥与功能微生物相结合制成微生物有机肥后施用,对土传病害有一定的防治作用,但效果不是太稳定。
藜麦作为抗逆性较强的新型杂粮作物,我国引进的时间较短,对其生理生化特性以及遗传机制了解还不够深入,面对产生的连作障碍问题,很难达到对症下药,但根际土壤是根际微生物直接作用于植物的重要场所,也是根际微生物受植物根系和分泌物影响最直接的区域,作物根际微生物能够分解并转化根际养分供给植物根系吸收,提高植物对生物和非生物胁迫的抵抗力[5-7]。夏围围等[8]比较了新一代高通量测序技术与传统的变性梯度凝胶电泳(Denaturing gradient gel electrophoresis,DGGE)指纹图谱技术,评价了这2种技术研究土壤微生物群落结构的优缺点,在不同的微生物分类水平,高通量测序草地土壤检测到22门、54纲、60目、131科、350属;而DGGE仅检测到6门、9纲、8目、10科、10属、表明DGGE显著低估了土壤微生物的群落组成。
新一代高通量测序技术的发展摆脱了研究土壤微生物分离培养的瓶颈,使得根际微生物和连作障碍之间的关系再次成为研究的热点。本试验采用新一代高通量测序技术,以静乐县娑婆乡连作藜麦试验地为研究对象,分析了第1年种植藜麦与重茬种植藜麦的根际土壤样品的细菌种群丰度和多样性的变化,并对细菌功能进行初步预测,试图探索藜麦连作障碍与土壤根际细菌之间的关系,以期了解藜麦连作对根际土壤细菌菌群多样性的影响,探究藜麦连作障碍机制,旨在从藜麦根际土壤微生物方面入手为藜麦的生产实践和可持续种植提供理论基础。
1 材料和方法
1.1 试验地概况
试验地选取我国藜麦之乡静乐县娑婆乡,于2015-2016年连续在同一地块进行试验。该地属于温带季风气候,夏季暖热且昼夜温差大,冬季寒冷,年降雨量380~500 mm,无霜期120~135 d。
1.2 试验方法
用无菌毛刷将藜麦根际土壤刷进无菌自封袋内,去除石块、大颗粒等杂质,同时在根周围用无菌铲取土壤放入无菌袋内,去除石块、大颗粒等杂质。每个重复取1个土壤样品,每个土壤样品取3个取样点然后混合。取样后立刻放入冰盒带回实验室,根际土壤放入-80 ℃冰箱用于DNA提取。
试验设2个处理,处理1为种植1 a的藜麦田(2016年),该处理设3次重复,编号分别为11,12,13,处理2为重茬种植的藜麦田(2015,2016年),该处理设3次重复,编号分别为C11、C12、C13。
1.3 土壤细菌的16S rRNA基因测序
1.3.1 DNA的提取及16SrRNA基因V4区片段的扩增 利用土壤DNA提取试剂盒提取6个样本的DNA,针对16SrRNA基因的V4区域进行PCR扩增预试验,然后进行大量PCR扩增试验。上游引物为530F(5′-AYTGGGYDTAAAGNG-3′);下游引物为805R(5′-TACNVGGGTATCTAATCC-3′);PCR反应体系为:Q5高保真DNA聚合酶:0.25 μL,反应缓冲液 5 μL,高GC缓冲液 5 μL,dNTP(10 mmol/L)0.5 μL,模板 DNA 1 μL,正向引物(10 μmol/L)1 μL,反向引物(10 μmol/L)1 μL,水11.25 μL;PCR扩增程序:98 ℃预变性2 min;98 ℃变性15 s,55 ℃退火30 s,72 ℃延伸30 s,共25个循环;最后72 ℃终延伸5 min结束。
1.3.2 PCR产物纯化与定量 对通过PCR聚合酶链式反应获得不同土壤样品的PCR产物后,进行琼脂糖凝胶电泳分析,针对目标条带进行割胶回收,获得纯化的PCR产物。
1.3.3 文库验证及测序 利用荧光分光光度计方法定量DNA,通过Agilent 2100对富集片段进行电泳完成质量控制,验证DNA文库片段大小及分布规律(Agilent 2100 bioanalyzer,Agilent 2100;Agilent High Sensitivity DNA Kit,Agilent, 5067-4626),然后送至上海派森诺生物科技有限公司,在Illumina-MiSeq平台上进行高通量测序。
1.4 数据处理与分析
为获得更为精准、高质量的生物学信息,首先对原始数据进行质量控制,获得最终用于分析的序列,然后应用QIIME软件,根据序列97%的相似度,将序列归并并划分为多个OUTs,并利用软件Mothur计算丰富度指数Chao1和ACE以及多样性指数Simpson和Shannon,并进行Alpha多样性分析,同时进行细菌群落分布、聚类分析和细菌功能预测分析。
2 结果与分析
2.1 不同处理土壤样本测序结果及质量控制
高通量测序结果显示,6个样本共获得有效序列276 400条(Sequences),运用QIIME软件(Quantitative Insights Into Microbial Ecology,v1.8.0,http://qiime.org/)剔除序列长度<150 bp的序列和 5′端引物错配碱基数> 1的序列以及含有连续相同碱基数>8的序列,然后调用USEARCH软件检查并剔除嵌合体序列。按照优化标准,去除不合格序列后共得到高质量序列251 346条,每个样本高质量序列占有效序列的比例都在89%以上(表1)。高质量序列用于样本间微生物丰富度和多样性的评估。
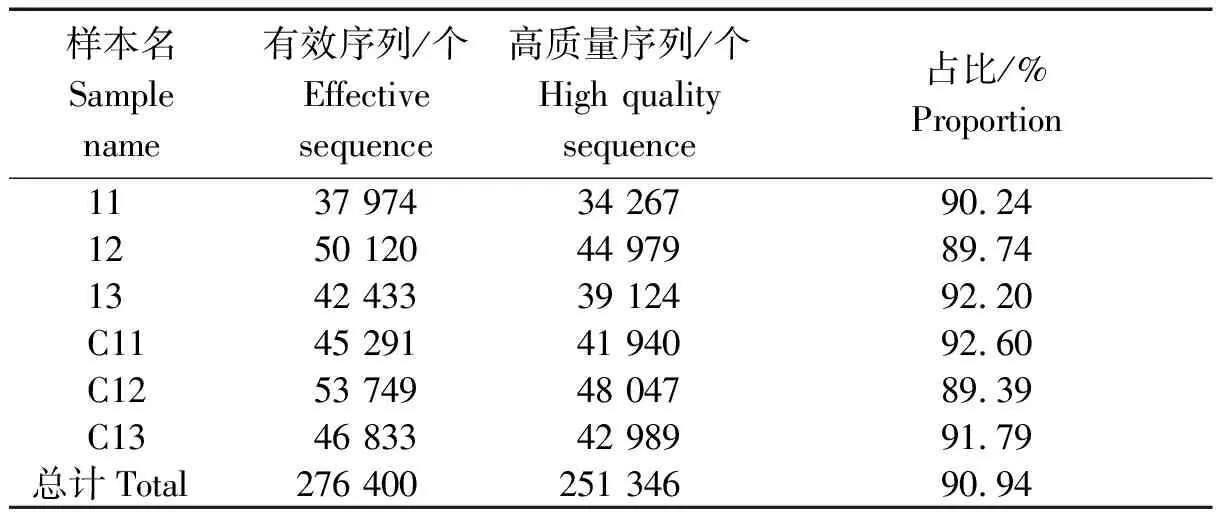
表1 各样本序列数据统计Tab.1 Statistics of sample sequences datas
稀疏曲线(Dilution curve)揭示了每个样品的取样深度,可以用来评价当前测序深度是否足以反映该群落样本所包含的微生物多样性。从图1可以看出,当测序量超过40 000读长时,仍然会有新的OUTs出现,但是由于曲线已经趋于平缓,表明该样品取样基本合理,在真实的环境中细菌群落结构的置信度比较高,能够比较真实地反映土壤中的细菌群落多样性,即当前测序深度足以反映该群落样本所包含的细菌群落多样性。
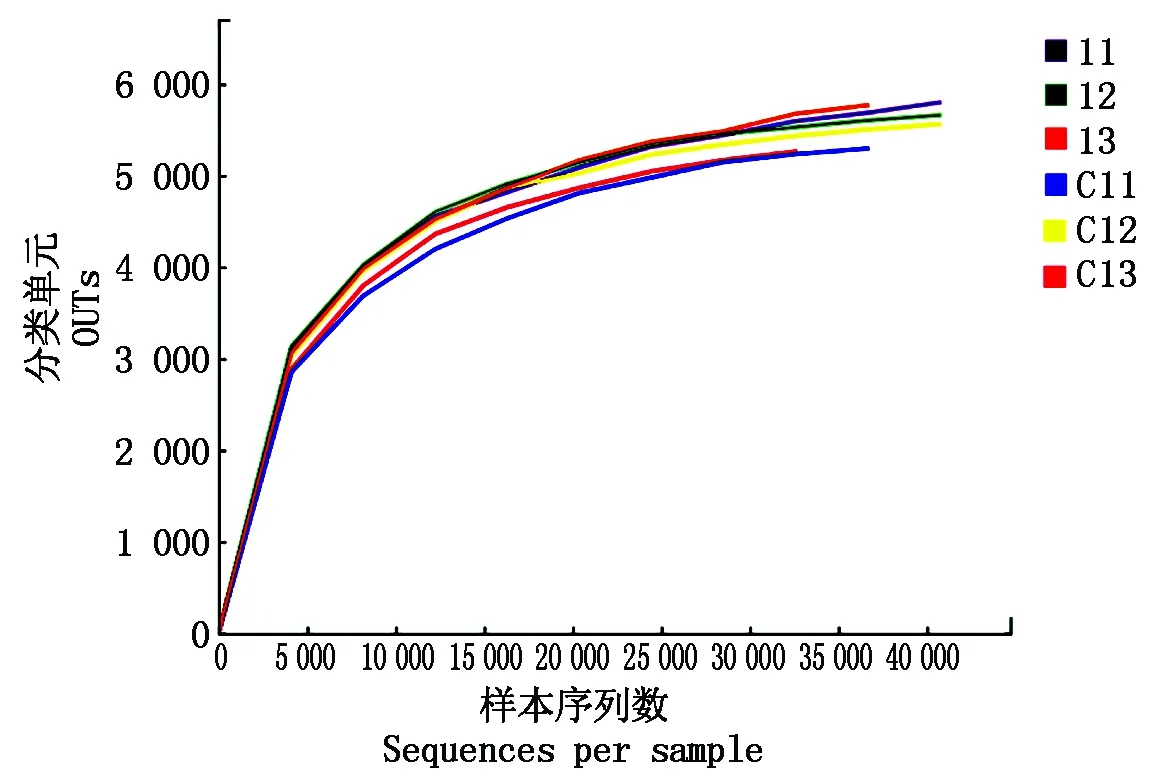
图1 各土壤样品的稀疏曲线(97%)Fig.1 Dilution curve of each soil sample(97%)
2.2 不同处理土壤细菌群落丰富度和多样性分析
利用Alpha多样性分析土壤中细菌群落的丰富度和多样性,各土壤样本的群落丰富度指数Chao1和ACE以及兼顾群落均匀度指数Shannon和Simpson如表2所示。
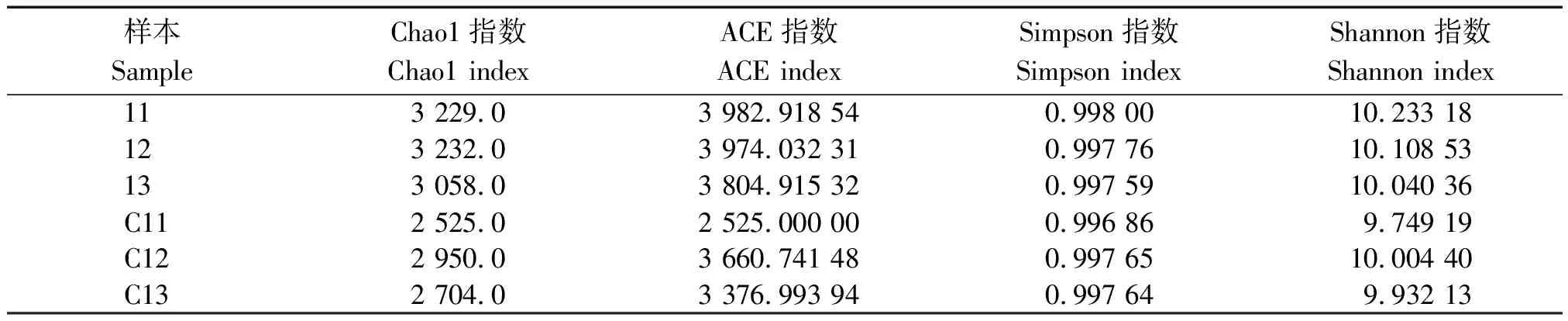
表2 土壤样本中细菌丰富度和多样性指数Tab.2 Index of the bacteria OUTs′ abundance and diversity in soil samples
就物种丰富度而言,种植1 a藜麦的土壤样本较种植2 a的土壤样本Chao1指数平均值增加16.4%,ACE指数平均值增加22.9%,表明种植2 a的土壤中细菌的物种丰富度大量减少,这与大部分研究结果一致。说明细菌物种丰富度减少可能是导致藜麦连作障碍的原因之一。 Simpson指数显示,种植1 a的土壤较种植2 a的土壤Simpson指数平均值增加0.4%,变化不明显,表明二者土壤中细菌群落的均匀度和优势OUTs数变化不明显;而Shannon指数显示,种植1 a的土壤较种植2 a的土壤Shannon指数平均值增加2.3%,表明种植1 a藜麦的土壤细菌群落多样性和稀有OUTs较种植2 a藜麦的土壤要高,说明重茬种植后不仅导致土壤细菌多样性下降,还导致部分稀有细菌种群多样性减少;土壤细菌在土壤养分循环的各个环节占有重要地位,其数量的下降和群落结构的变化均会引起微生物功能的失调,进而导致土壤养分和肥力的下降,这与徐雪雪[9]的研究结果基本一致。
2.3 不同处理土壤菌群的分类学组成分析
将各分类水平的群落组成数据根据分类单元的丰度分布或样本间的相似程度加以聚类分析并绘制热图(图2),种植1 a藜麦的土壤样品和种植2 a藜麦的土壤样品分别聚类在不同的类别,说明样品一致性较好,有一定的代表性。聚类结果表明,重茬种植后伦茨氏菌属(Lentzea)、溶杆菌属(Lysobacter)、中慢生根瘤菌属(Mesorhizobium)、极地单胞菌属(Polaromonas)多样性增多;分枝杆菌属(Mycobacterium)、藤黄单胞菌属(Luteimonas)、芽单胞菌属(Gemmatimonas)、浮霉菌属(Planctomyces)等细菌多样性减少。增多和减少的细菌种群分属不同的类别。其中,藤黄单胞菌属细菌为革兰氏阴性、好氧的棒状杆菌,目前共有10个种。李广宁等[10]分离出1株藤黄单胞菌属细菌 HF-1127,通过研究该菌株的形态、培养特征、生理生化特征和遗传特性,表明该细菌具有抗菌活性,其代谢产物对大肠杆菌和金黄色葡萄球菌有抗性;范晓阳[11]从海水中分离出一种深渊藤黄单胞菌,研究表明,其可以降解明胶、淀粉、吐温-20、吐温-40以及吐温-80等物质;黄佩蓓等[12]对浮霉状菌研究认为,浮霉状菌属细菌是一类具有重要生态作用的环境微生物,参与环境中的碳和氮的循环。
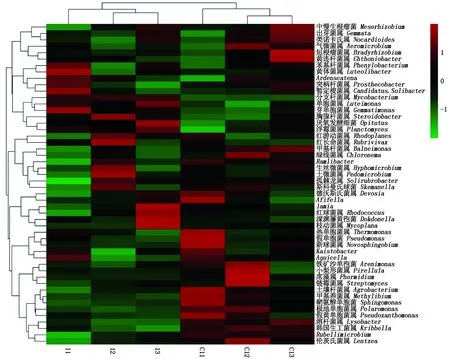
图2 结合聚类分析的属水平群落组成热图Fig.2 Heat map of the community composition in genus level combined with the cluster analysis
2.4 不同处理土壤样品序列的菌群代谢功能预测
大量研究表明,土壤中的群落微生物功能组成与环境因子的相关度要大于物种组成与环境因子的相关度,在相似的环境条件下,虽然微生物群落的功能相似,但是行使功能的微生物组成差异反而较大,因此,在揭示土壤环境中的微生物群落组成的基础上,最终要揭示微生物群落的功能。运用PICRUSt软件通过将现有的群落组成数据与KEGG数据库中微生物代谢功能的类别对比进行群落样本功能预测[13]。由图3可知,重茬种植后,行使环境信息处理功能类别中编码细胞膜运输的功能基因大量减少;行使遗传信息处理功能类别中编码翻译、复制和修复的功能基因大量减少;行使代谢功能类别中编码异生素生物降解和代谢、聚糖生物合成和代谢的功能基因大量减少,编码核苷酸代谢、萜类化合物和聚酮化合物的代谢、辅因子和维生素的代谢等功能基因少量减少;编码信号转导和脂质代谢的功能基因有少量增加。

图3 群落样本的代谢功能预测图Fig.3 Prediction map of the metabolic function in community samples
3 结论与讨论
作物连作障碍产生的原因多且比较复杂,其中,土壤微生物丰富度和多样性的变化是导致连作的主要原因之一。在农业生态系统中,土壤微生物多样性对维持生态系统的平衡具有重要作用。由于技术的限制,早期对土壤微生物研究需要依赖于人工分离培养,而大部分土壤微生物无法在实验室条件生存,因此阻碍了人们对微生物的了解,但是随着技术的进步,尤其是下一代高通量测序技术的发展,不再依赖人工培养,使研究者们能够同时对多种微生物基因组进行测序,通过对土壤根际微生物的丰富度和多样性的研究有助于更好地研究微生物与作物生长发育的关系,为克服作物连作障碍提供数据支撑[14]。
藜麦重茬种植会导致根际土壤的细菌种群数量和多样性都有不同程度的降低,同时也导致致病菌数量的增多,使根际土壤细菌种群朝着不利于植物生长的方向发展,这与大量作物连作障碍研究结果基本一致[15]。Bever等[16]根据多年的研究结果提出了土壤-植物反馈机制,该机制根据最终的结果又分为正反馈、负反馈和中性反馈。连作障碍的发生就是由于土壤与植物连续负反馈的结果。同时藜麦重茬种植后细菌种群结构也发生了变化,伦茨氏菌属、溶杆菌属、中慢生根瘤菌属、丛毛单菌属等细菌数量增多;分枝杆菌属、藤黄单胞菌属、芽单胞菌属浮霉菌属等细菌数量减少。据报道,溶杆菌属细菌对线虫和植物病害(革兰氏阴性菌、革兰氏阳性细菌、真菌等)具有生防作用,同样具有固氮作用的中慢生根瘤菌属细菌数量也有增加[17]。但是由于相关藜麦连作障碍方面的研究尚未见报道,因此可能是由于藜麦根际微环境比较适合溶杆菌属细菌以及中慢生根瘤菌属细菌的繁殖,导致了其数量的增加;但是参与土壤中碳和氮循环的浮霉菌属细菌数量却降低了,藜麦连作打破了藜麦根际土壤细菌之间的平衡,这可能导致藜麦根际土壤细菌功能性紊乱,从而负反馈于植株体本身,导致了藜麦的连作障碍,但具体的作用机制和相互之间的关系还需要进一步研究。
土壤根际微生物群落结构的差异会表现在基因表达方面的差异,藜麦重茬根际土壤中编辑复制和修复以及外源性物质降解和代谢的功能基因大量减少,受环境条件的影响越来越大,导致细菌对外来入侵的抵抗能力大大减小,原来有益的细菌群落丰度显著下降,病原菌细菌群落大量增殖,因此,导致重茬种植藜麦时生长受阻,土传病害增加,产量急剧下降。也有研究表明[18],连作不仅仅会导致有益细菌群落丰度降低,有益真菌的数量也会大幅度降低,而病原菌真菌的数量则会大幅度增加。由于藜麦连作障碍土壤根际细菌多样性方面的研究尚未见报道,并且本研究所采用的高通量测序技术的第2代测序技术,对细菌的分类仅限于属的水平,无法定位到某一种细菌,且同一属的不同细菌种在土壤中可能会行使不同的功能,而不同属的细菌也可能在土壤中行使同样的功能,研究所得到的结果可能会有偏差。而随着第3代测序技术(单分子测序技术)的进一步完善和发展,打开了一扇精准研究土壤微生物的大门,该技术已经能够将土壤微生物定位到种的水平,对根际微生物的研究将会更加全面和细化,现在无法解决的问题可能会在不久的将来得到解决。
本研究中,利用高通量测序技术对2个处理6个样本的土壤微生物进行了16SrRNA基因的V4区测序,然后对测序数据进行了处理和分析,该项研究的样本量和测序深度在藜麦的相关研究中尚属首次,能够更加全面地揭示许多利用传统分子生物学技术研究土壤微生物无法解决的问题,结果中获得的土壤微生物多样性变化基本上能够揭示藜麦连作后土壤细菌种群多样性的变化,并且预测了土壤细菌种群代谢功能的变化,为今后研究藜麦连作障碍机制提供参考,但由于本研究中测序深度即稀释曲线并没有达到完全饱和,处理中还有部分细菌种群没有被发现。因此,本研究结果仅代表各个土壤样品中的大部分细菌种群,今后仍有待开展更深层次的研究。
