嵌合抗原受体 T细胞治疗卵巢癌的抗原靶点研究进展△
2019-04-28李浩兰李桐李文忠路国秋王建东
李浩兰,李桐,李文忠,路国秋,王建东
1北京中捭生物科技有限公司,北京100080
2首都医科大学附属北京妇产医院妇瘤科,北京100010
卵巢癌(ovarian cancer,OC)是致死率最高的妇科恶性肿瘤,由于发病隐匿,缺乏有效的早期诊断方法,约70%的患者在就诊时已属于晚期[1],经过手术以及化疗后仍有超过80%的患者会复发[2]。寻找新的OC治疗方法对于提高患者的生存率具有重要意义。嵌合抗原受体T细胞(chimeric antigen receptor T cell,CAR-T)治疗技术是新型的抗肿瘤免疫疗法,在血液肿瘤治疗中取得了很好的效果,但在OC中应用较少,其治疗效果和OC的抗原靶点有关。本文回顾和总结CAR-T技术以及卵巢中可应用于CAR-T治疗的不同类型的靶点抗原,旨在为卵巢癌CAR-T治疗的靶点选择提供理论支持。
1 CAR-T技术
CAR-T是一种经过基因修饰来靶向肿瘤特异性抗原的自体T细胞疗法,现已经开发到第四代[3]。第一代嵌合抗原受体(chimeric antigen receptor,CAR)结构在2006年首次被构建,其包含细胞外识别肿瘤抗原的单链可变区(single-chain variable fragment,scFv),促进CAR和与靶细胞表面膜近侧抗原结合的铰链区以及细胞内的免疫细胞活化序列CD3ζ。第一代CAR-T在OC的Ⅰ期临床试验中并未显示出明显的抗肿瘤作用[4],故研究者开始对CAR的结构进行改造以增强CAR-T的抗肿瘤作用。第二代CAR在第一代的基础上增加了一个细胞内共刺激信号元件,如 CD28、CD134、CD137或 CD27[5]。共刺激信号元件的加入显著延长了CAR-T在体内的存活时间并增强了其抗肿瘤作用,首次用于B细胞淋巴瘤治疗也展示出了良好的抗肿瘤效果[6]。第三代CAR则包含两个及以上的共刺激信号元件,旨在提高转基因的表达,促进CAR-T的激活和扩增,在体内试验中包含CD28和CD137两个共刺激元件的第三代CAR-T显示出更强的抗肿瘤活性[7]。第四代CAR则加入了细胞因子来增强CART杀伤肿瘤细胞的效果[8]。经过十多年的发展,CAR结构基本明确:包含细胞外特异性识别肿瘤抗原的scFv片段、促进与肿瘤表面膜近侧抗原结合的铰链区、固定CAR的跨膜区、增强T细胞活化的共刺激元件和T细胞活化信号转导CD3ζ链[9]。当靶细胞上的抗原与CAR上的受体结构域结合时,通过铰链区和跨膜区将信号传递至细胞内,细胞内信号域将信号转化为活化信号,激活效应细胞进行增殖,产生细胞因子和穿孔素等杀伤靶细胞[10]。目前多采用第二代(单共刺激信号元件)和第三代(双共刺激信号元件)CAR进行临床试验。(图1)

图1 四代CAR的结构示意图
CAR-T疗法已经在血液肿瘤治疗中取得了突破性成就。第一个靶向CD19的CAR-T药物CTL019用于治疗儿童及年轻成人复发性/难治性急性淋巴细胞白血病,3个月完全缓解率可达83%[11],并于2017年8月获得美国食品药品管理局(FDA)批准上市。此外,另一药物YESCARTA在治疗大B细胞淋巴瘤时完全缓解率可达51%[12],并最终于2017年10月获得FDA批准上市,成为首款针对特定非霍奇金淋巴瘤的CAR-T治疗药物。CAR-T在实体瘤的治疗中也展现出了巨大的潜力。2016年12月,希望之城(Hope of City)研究中心利用靶向白细胞介素-3 受体 α2(interleukin-3 receptor α2,IL-3Rα2)的CAR-T治疗复发性多灶胶质母细胞瘤,10次脑室注射治疗后,患者肿瘤病灶完全消失且未出现强烈的不良反应[13]。2017年7月宾夕法尼亚大学关于靶向表皮生长因子受体Ⅲ型突变体(epidermal growth factor receptor variantⅢ,EGFRVⅢ)阳性神经胶质瘤的CAR-TⅠ期临床试验结果表明,CAR-T能够成功穿越血脑屏障,在肿瘤位点发现了CAR-T的浸润[14]。此外,针对肺癌、结直肠癌、胰腺癌等肿瘤均已开展靶向特异性肿瘤抗原的CAR-T疗法[15-17]。
2 CAR- T治疗OC的抗原靶点
目前CAR-T疗法面临的主要挑战是临床安全性,因此选择合适的靶点对于CAR-T疗法的安全性和有效性至关重要。理想的肿瘤靶点应该在恶性肿瘤组织中表达而在正常组织中不表达,因为即使在正常组织中只有低剂量的表达也会导致急性和严重的脱靶毒性[18-20]。目前国内针对OC的CAR-T治疗的研究很少,但国际上一些著名研究机构如纪念斯隆凯瑟琳癌症中心(Memorial Sloan-Kettering Cancer Center,MSKCC)和宾夕法尼亚大学等已经开展了关于OC的CAR-T治疗研究(表1)[1,4,15,21-27]。
2.1 人表皮生长因子受体(human epithelial growth factor receptor 2,HER 2)
HER2为人表皮生长因子受体家族的成员之一,是一种具有酪氨酸激酶活性的跨膜受体样蛋白,分子量为185 kD,由与配体结合的胞外区、信号转导的跨膜区和具有激酶活性的胞内区组成[1]。HER2能够抑制细胞凋亡,诱导新生血管生成,提高细胞运动能力从而促进肿瘤的生长增殖和浸润转移[28]。HER2在正常组织如胃肠道、呼吸道、尿道、皮肤、乳腺和胎盘中均有不同程度的表达[29]。研究表明,在2%~66%的OC组织中能够检测到HER2的过表达[30],因此HER2可作为OC CAR-T治疗中的特异性靶点。晚期OC患者通常高表达HER2,并且患者体内可以检测到CD4+和CD8+T细胞的响应[31]。目前针对OC的CAR-T研究已完成临床前体外实验,可证明HER2-CAR-T能够识别并杀伤表达HER2的OC细胞,而对HER2表达阴性的细胞不产生响应[32],现已有两项Ⅰ期临床试验正在进行中。
2.2 间皮素(mesothelin,MSLN )
MSLN是一种糖基磷脂酰肌醇(glycosylphosphatidylinositol,GPI)锚定蛋白,分子量为69 kD,蛋白酶加工后形成一个40 kD的膜结合蛋白和一个31 kD称之为巨核细胞促进因子的脱落片段并释放至细胞外[33]。MSLN能够通过影响丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)/细胞外调节蛋白激酶(extracellular regulated protein kinases,ERK)和磷酸肌醇3-激酶(phosphatidylinositol 3-kinase,PI3K)通路参与调节细胞的凋亡,同时还参与信号转导及转录激活因子3(signal transducer and activator of transcription 3,STAT3)/核因子-κB(nuclear factor-κB,NF-κB)通路来影响细胞的增殖,进而促进肿瘤的发生和发展[34]。MSLN一般限制性地表达于胸膜、腹膜、心包的浆膜细胞及气管、卵巢、扁桃体、输卵管的间皮细胞上。研究证明,MSLN表达于多种实体瘤中,其中约70%的OC组织中存在MSLN的表达[35],因此MSLN可作为OC CAR-T治疗的潜在靶点。临床前实验结果表明,人源化MSLN-CAR-T在体外与MSLN阳性细胞共培养时能够产生炎性因子并有效裂解靶细胞,体内回输OC细胞移植小鼠模型表现出一定的抗肿瘤效应[21],同时并未出现脱靶毒性。目前已有两个靶向MSLN的CAR-T临床试验分别在美国(已完成)和中国展开(正在招募中)。
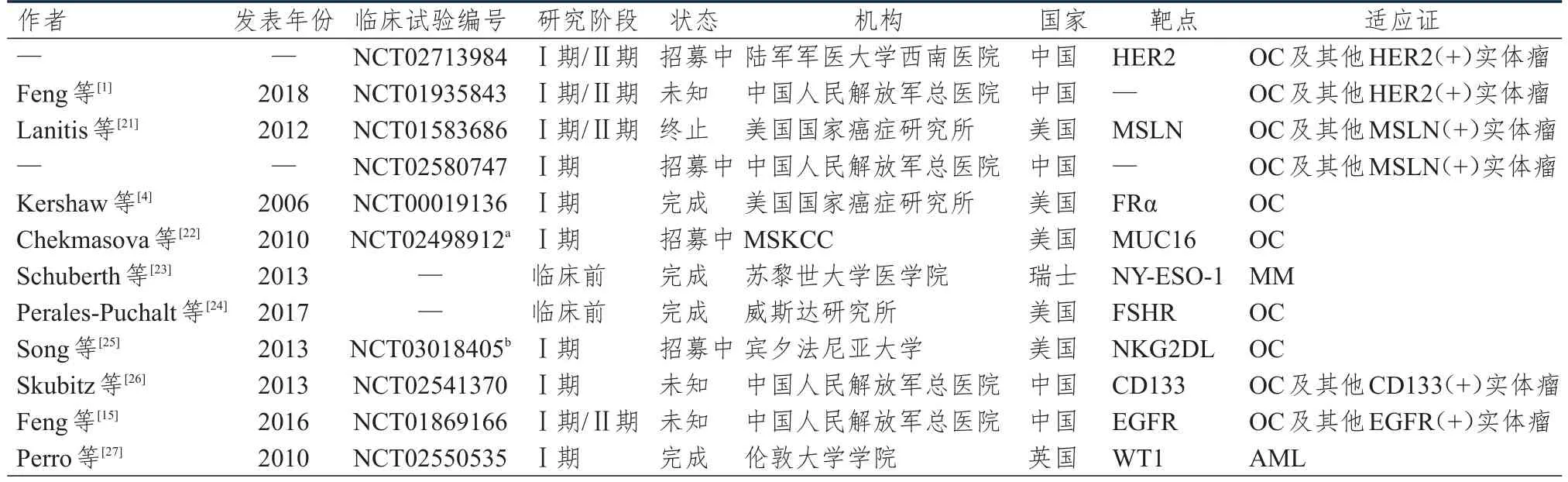
表1 CAR-T疗法靶向特异性肿瘤抗原治疗OC的试验总结
2.3 叶酸受体 α(folate receptor α,FR α)
FRα是由GPI锚定的一个38 kD的蛋白,介导叶酸的摄入、细胞增殖、氨基酸代谢、DNA和RNA的合成及甲基化等过程[30]。FRα在肾、胃、肺等正常组织的上皮细胞腔面上有微弱的表达,在多种肿瘤组织如OC、子宫内膜癌、非小细胞肺癌、结肠癌、乳腺癌中过表达。其中超过90%的OC组织高表达FRα,表达水平比正常卵巢组织,高90倍[36]。此外,考虑到FRα的生物学功能,FRα的CAR-T治疗将会是相对安全和易耐受的,这使得FRα成为OC CAR-T治疗的理想靶点。临床前实验结果证明,加入共刺激元件CD27的第二代FRα-CAR-T展示出了有效的裂解OC细胞的能力[4]。目前针对FRα-CAR-T治疗的Ⅰ期临床试验已完成。
2.4 黏蛋白 16(mucin 16,MUC16)
MUC16是一种高度糖基化的Ⅰ型跨膜糖蛋白,由60个重复序列的156个氨基酸的胞外段、跨膜段和32个氨基酸的胞内段组成。MUC16参与调节细胞的代谢和增殖过程[37]。MUC16能够抑制自然杀伤(natural killer,NK)细胞的激活从而阻止NK细胞与肿瘤细胞结合,NK细胞也能够与MSLN结合,提高细胞黏附性,同时MUC16还可以与细胞质内JAK激酶2(Janus kinase 2,JAK2)结合上调细胞周期蛋白D1(cyclin D1)和胚胎干细胞多能性维持关键基因(Nanog homeobox,NANOG)的表达,从而促进肿瘤的发生和发展[38]。MUC16在正常组织中几乎不表达,而在超过80%的OC细胞中高表达,另外在子宫内膜癌、胰腺癌、结肠癌、乳腺癌和肺癌细胞中也有过表达[39]。MUC16在肿瘤细胞中存在剪切机制,胞外段糖类抗原 125(carbohydrate antigen 125,CA125)经剪切后被释放,并可在血浆中被检测到。CA125作为OC免疫标志物检测中的重要参考指标,超过90%的OC患者血清中CA125的表达上调,而术后患者体内CA125高表达者复发率高达85%~93%[40]。因此MUC16同样可作为OC CAR-T治疗的理想靶点。MSKCC针对MUC16开发的单靶点CAR-T在临床前研究中表现出较好的抗肿瘤效果,现正开展Ⅰ期临床研究[22]。
2.5 纽约食管鳞状细胞癌 1(New York esophageal 1,NY-ESO- 1)
NY-ESO-1属于癌-睾丸抗原(cancer-testis antigen,CTA)家族,分子量为18 kD,N端含有甘氨酸富集区,C端为一个疏水氨基酸尾,NY-ESO-1的作用机制尚不清楚[41]。作为CTA,NY-ESO-1在正常组织中仅限制性地表达于睾丸和胚胎组织,但是在多种肿瘤组织,如OC、神经母细胞瘤、黑色素瘤等中均有高表达[42]。此外,由于血睾屏障的存在,正常组织并不会发生免疫毒性,因此NY-ESO-1是OC CAR-T治疗的理想靶点。研究发现,约43%的OC肿瘤组织中高表达NY-ESO-1,并且能够对NYESO-1多肽发生响应从而产生CD8+T细胞[43],并且这种CD8+T细胞能够在71%的OC患者的肿瘤浸润淋巴细胞和肿瘤相关淋巴细胞中检测到[44]。目前针对NY-ESO-1的CAR-T治疗在OC中未开展相关研究,但对于多发性骨髓瘤治疗已完成临床前阶段,第二代NY-ESO-1-CAR-T细胞在体外培养过程中能够释放炎性因子并有效裂解靶细胞,同时也能够发挥明显的体内抗肿瘤作用[23]。目前针对表达NY-ESO-1的OC的临床试验多采用T细胞抗原受体(T cell receptor,TCR)疗法,且未见不良反应[45]。这为NY-ESO-1的CAR-T治疗的安全性奠定了基础。
2.6 卵泡刺激激素受体(follicle stimulating hormone receptor,FSHR)
FSHR是由寡聚糖蛋白构成的七次跨膜受体,属于G蛋白偶联受体家族成员,由695个氨基酸组成[46]。研究发现,FSHR能够通过与卵泡刺激素结合来调节性腺的发育,并在肿瘤血管形成中发挥作用[47]。FSHR仅在睾丸支持细胞和卵巢粒层细胞中表达,而在其他正常组织中不表达。研究发现,FSHR在33%的透明细胞性OC、67%的黏液性OC和70%的子宫内膜癌中均存在高表达[48],因此,FSHR可作为OC CAR-T治疗的潜在靶点。目前针对OC靶向FSHR的CAR-T治疗已经完成了临床前试验,结果显示FSHR-CAR-T细胞能够特异性杀伤表达FSHR的OC细胞,同时在体内试验时表现出明显的抗肿瘤效果,显著地提高小鼠的生存率,并且没有观察到不良反应[24]。
2.7 其他抗原
除了上文提到的一些抗原,还有其他一些潜在靶点,如自然杀伤细胞2族成员D配体(natural killer group 2 member D ligand,NKG2DL)、CD133、表皮生长因子受体(epidermal growth factor receptor,EGFR)、肾母细胞瘤1(Wilms tumor 1,WT1)等,在OC中也呈现较高的表达水平。
NKG2DL广泛表达于OC组织中[49]。目前针对OC的NKG2DL-CAR-T治疗已经完成临床前试验,NKG2DL-CAR-T细胞能够有效杀伤表达NKG2DL的OC细胞[25]。Ⅰ期临床试验已经展开。
CD133为一个干细胞标志物抗原,在约31%的OC组织中有表达[50]。临床前实验中,CD133-CART细胞能够有效发挥抑制OC移植瘤小鼠肿瘤生长的作用[26],靶向CD133治疗OC的CAR-T治疗也已经开展。
EGFR和HER2同属于人表皮生长因子受体家族的成员,能够促进肿瘤血管的生成。EGFR在35%~70%的OC中有表达[51]。临床前试验表明EFGRCAR-T细胞能够有效杀伤表达EGFR的非小细胞肺癌细胞[15],针对高表达EGFR的OC的CAR-T临床试验也在进行中。
WT1能够促进肿瘤的浸润和转移,在超过50%的OC和子宫内膜癌中呈现高表达,且WT1高表达的肿瘤患者通常预后较差[30]。目前以WT1为靶点的OC治疗已经展开了TCR治疗研究[27],这为WT1的CAR-T治疗奠定了基础。
3 小结与展望
CAR-T疗法经过近几年爆炸式的发展,已经成功应用于血液病以及部分实体肿瘤的治疗,并与“免疫检查点阻断抗体疗法”一起被评为“2013年度十大科学突破”之首[52]。目前针对OC的CAR-T疗法在临床前试验和临床试验中均表现出较好的治疗效果,但也存在着一些限制。首先,临床上,CART回输后可能引起包括脱靶效应、细胞因子释放综合征、发热反应等不良反应,因此选择合适的靶点抗原能够有效避免脱靶效应,使CAR-T发挥最大作用。此外,由于高度异质化的实体肿瘤不会均一地表达单一靶抗原,且单一靶点的CAR-T治疗往往会诱发对应靶抗原的丢失,使得单一靶点的CAR-T不能有效识别和杀伤肿瘤细胞,从而限制其治疗效果。目前已有研究组织利用双靶点技术同时靶向多个抗原[53-54],这样能够有效避免抗原的逃逸,也提高了CAR-T治疗的安全性。其次,肿瘤微环境中存在的免疫负调控机制会抑制肿瘤微环境内T细胞的抗肿瘤免疫反应,导致T细胞耗竭,影响CAR-T的治疗效果。目前已有多个研究组织和机构将CART技术同免疫检查点抑制剂联用,一方面解除T细胞的免疫抑制状态,一方面增强T细胞对肿瘤细胞的攻击力,从而对OC发挥更强的疗效。
将CAR-T技术应用到OC的治疗中,并通过选择合适的抗原、双靶点技术和同免疫检查点抑制剂联用等方法解除CAR-T技术在OC临床治疗中的限制,不仅能够丰富现有有限的治疗手段,同时有望突破OC复发率高、效果不佳的困境,达到“根治”的效果,从而使更多的患者从中受益。
