硫原子在镍基合金825(001)面吸附的第一性原理研究
2019-04-28黄泰愚刘建仪
范 舟, 黄泰愚, 黄 晋, 刘建仪
(1.西南石油大学,材料科学与工程学院, 成都 610500; 2.西南石油大学,油气藏地质及开发工程国家重点实验室, 成都 610500)
1 引 言
目前对于S和SO2,H2S等含硫化合物的吸附研究受到国内外的广泛关注[1,2].主要原因有:一方面, 在很多需要过渡金属催化的化工业中, 即使存在很少量的硫吸附也会使催化剂中毒,降低过渡金属的催化作用[3];另一方面, 在石油天然气开采输送和炼油化工等领域,硫的局部聚集吸附, 会使得输送管线发生局部腐蚀,这类腐蚀会在事前没有明显征兆条件下造成灾难性后果, 造成巨大经济人员损失[4]. 因此研究S和SO2、H2S吸附在金属表面的相互作用关系, 对于防止和降低催化剂中毒以及抗硫腐蚀的新材料和缓蚀剂的研发有重要意义.
目前在实验方面关于硫及其化合物在金属表面吸附作用的报道甚多[5-9], 研究表明:S和H2S吸附在钢铁等金属表面进行腐蚀过程中,腐蚀程度和腐蚀产物与环境密切相关, H2S吸附在Fe表面硫化过程中, 100 K以下低温时以分子形式存在, 中高温H2S会发生分裂, 不再是原来的三角分子结构. 实验手段对S和SO2、H2S等含硫化合物的吸附研究比较局限, 很难对吸附后的电子结构和成键类型、成键强弱作进一步的研究, 因此有必要利用第一性原理计算方法从电子层面研究吸附的微观机理和本质特征[10-15].
为了满足高温高压, 高含硫油气井的开采需求, 油套管必须采用高合金不锈钢和镍基合金. 对于S腐蚀镍基合金的研究, 目前[16]已经通过SEM、EDS等技术方法研究了S在镍基合金825表面吸附后的腐蚀产物形貌和成分,然而, 对于S在镍基合金825这类被广泛应用于高温高压苛刻腐蚀环境中合金[17]的微观吸附研究却未曾涉及. 因此本文采用密度泛函理论方法研究了S原子在镍基合金825(001)面的吸附, 计算了吸附能, 吸附位置, 态密度, 差分电荷密度, 分析了合金表面的弛豫情况, 从而有助于透彻得认识S吸附在镍基合金825(001)面的微观电子结构和作用机理, 为防止和减少油气田管道硫腐蚀的研究提供理论指导.
2 计算模型及方法
本文的所有计算均采用第一性原理软件Material Studio中的castep模块[18]计算完成. 计算采用广义梯度近似(GGA)中的PBE泛函[19], 计算体系中核外电子和原子核之间的作用均采用超软赝势[20]进行处理,自洽运算总能量收敛为1.0×1.0-5eV/atom, 原子间相互作用力收敛于0.02 eV/Å, 应力偏差小于0.05 GPa, 公差偏移小于0.002 Å. 在确立了镍基合金825最优结构后, 合金825(001)面采用五层金属原子构成的平面模板进行模拟, 为了避免平板之间的相互作用, 在平板之间添加了12 Å的真空区域. 五层平板模型中, 下面两层固定不动作为基底体层,上面三层作为表面, 在结构优化中跟吸附原子分子进行自由弛豫. 上述所有参数均通过改变k点取样和截断能等参数进行了收敛性测试, 结果表明以上参数能保证计算精确度.
3 计算结果与分析
3.1 镍基合金825晶胞结构优化
镍基合金825(以下简称合金825)主要是由Ni, Cr, Fe三种金属元素组成, 通过相关资料查阅可以得到, 其主要成分为:Ni含量38%-46%(质量分数, Cr含量19.5%-23.5%, 还有少量微量元素, 余量是铁. 化学元素成分如表1所示.

表1 合金825元素成分表(wt. %)
合金825[21]是一种添加少量微量元素的镍-铁-铬固溶强化镍基耐蚀合金. 在合金825中, 溶质元素(Cr、Fe)和溶剂元素(Ni)在原子半径和电负性方面相似, 因此合金825容易形成替换固溶体而非间隙固溶体. 由表一可以得到, Ni, Cr和Fe原子的比例为2:1:1, 根据替换固溶体我们可以得到三种可能的晶胞结构, 如图1所示.
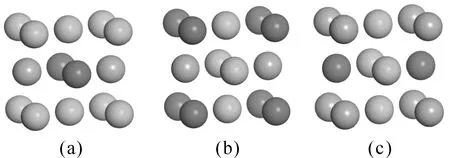
图1 合金825三种可能的高对称晶胞结构(图中绿色为Ni原子,红色为Cr原子, 紫色为Fe原子)Fig. 1 Nickel-based alloy 825 with three possible unit cells (Ni atom-green, Cr atom-red, Fe atom-purple).
为了获得Ni基合金825最合理结构, 表2列出了三种合金晶胞几何优化后的晶格常数, 并与XRD实验PDF卡片晶格常数值进行了对比. 相比较可以看出,结构A误差最小, 与实验合金825晶格常数最为接近.
晶胞的总能量越低, 晶胞结构则越稳定[22], 因此我们计算了三种晶胞结构的总能量(见表3). 结构A即Ni原子占据顶角位置要比B, C结构能量更低, 说明Ni原子优先占据面心立方的顶点位置, 这与表2晶格常数误差分析相佐证, 因此可以得到结构A为合金825最稳定晶胞结构. 因此S原子在合金825表面的吸附模型以A晶胞结构为基础进行建立.
表2 三种晶胞结构几何优化后的晶格常数
Table 2 Lattice constants of the three cell structures after geometrical optimization
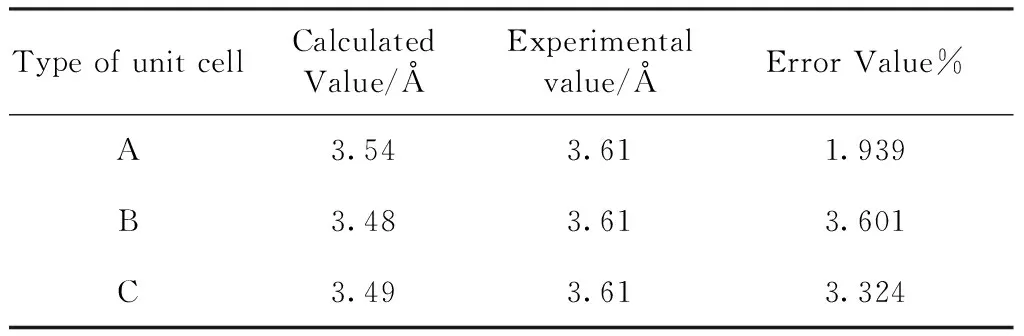
Type of unit cellCalculatedValue/ÅExperimentalvalue/ÅError Value%A3.543.611.939B3.483.613.601C3.493.613.324
表3 三种晶胞结构的总能量
Table 3 Lattice energy of the three structure after geometrical optimization
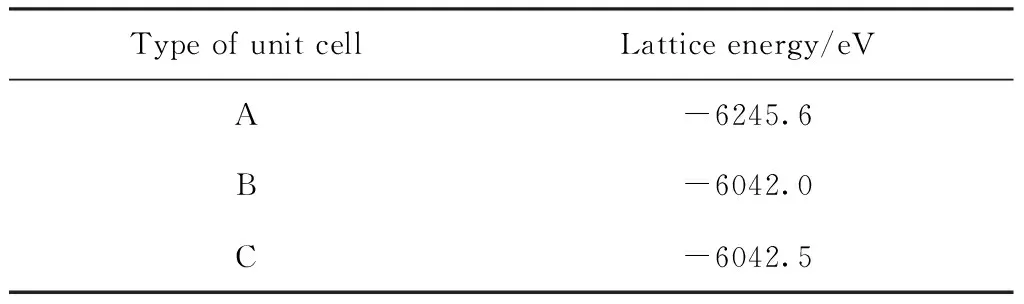
Type of unit cellLattice energy/eVA-6245.6B-6042.0C-6042.5
3.2 S原子在合金825(001)面的吸附
3.2.1吸附结构和吸附能
合金825是一种典型的面心立方(FCC)合金, 在三个典型的低指数晶面中对点蚀最敏感的表面是(001)面, 而耐腐蚀性较强的晶面是(111)和 (101)面[23]. 因此, 我们研究了合金825的点蚀敏感面(001)面作为S原子的吸附面. 本文采用3.1获得的最优合金825结构的P(1×1)原胞进行结构优化, 然后采用P(2×2)周期性超胞作为初始表面讨论了0.25ML覆盖度下S原子在合金825(001)面Ni-Fe终止面和Ni-Cr终止面的吸附情况, 如图2所示(图中, 黄色的为S原子, 绿色为Ni原子, 红色紫色分别为Cr原子和Fe原子).

图2 S原子在合金825(001)面的初始吸附构型 (图中T表示顶位(Top), B表示桥位(Bridge), H表示四重穴位(Hollow)).Fig. 2 Initial adsorption structure of S on 825 alloy (001) surface. (T-Top site, B-Bridge site, H-Hollow site)
为了分析S原子在不同吸附位置的稳定性, 我们计算了S原子的吸附能, 吸附能计算公式[14]如下:
Ead(S)=ES/Ni825-Slab-ENi825-Slab-ES
(1)
其中,ES/Ni825-Slab,ENi825-Slab,ES分别为吸附S原子后的合金Slab结构总能, 吸附前的合金Slab结构总能, 孤立S原子的能量. 由定义可知, 吸附过程为放热反应, 吸附能为负值, 数值绝对值越大, 放热越多, 说明结构越稳定.
我们计算了S原子在合金825(001)面上所有高对称位置(S最倾向吸附的位置)的吸附能(如表4). 结果显示, S原子在 Ni-Fe终止面和Ni-Cr终止面的各个高对称位置吸附能均为负, 说明S在合金表面是主动进行的放热反应, 且绝对值越大, 说明放热越多, 吸附作用也就越强. 比较吸附能的大小可以得到, S在Ni/Fe 终止面的吸附能明显大于在Ni/Cr终止面上的吸附能, 说明与Ni/Cr终止面相比, S更倾向于吸附在合金825(001)面Ni/Fe面上. 对比S在Ni/Fe面上各个高对称位置的吸附能也可以看出, S最倾向吸附于Ni/Fe终止面上的四重穴位, 这与S在Fe(001)面的吸附结果相类似[14]. 吸附过程中, 我们发现原子S的初始位置在Ni顶位和Fe顶位时, 它最终会慢慢向四重穴位偏移, 而初始吸附位在Cr顶位时, S原子则比较稳定, 随着吸附的发生, 偏向并不明显; 当S的初始吸附位为桥位时, 除了Cr-Cr桥位比较稳定, 其他高对称桥位均发生了比较明显的向四重穴位偏移的趋势, 偏向Ni/Fe面四重穴位的倾向尤其明显. 结合吸附能与吸附过程中S原子的倾向偏移, 不难发现Ni/Fe终止面上的四重穴位是S原子吸附在合金825(001)面上的最稳定吸附位置, 因此我们后续重点进行S在Ni/Fe终止面以及其面上穴位的吸附研究.
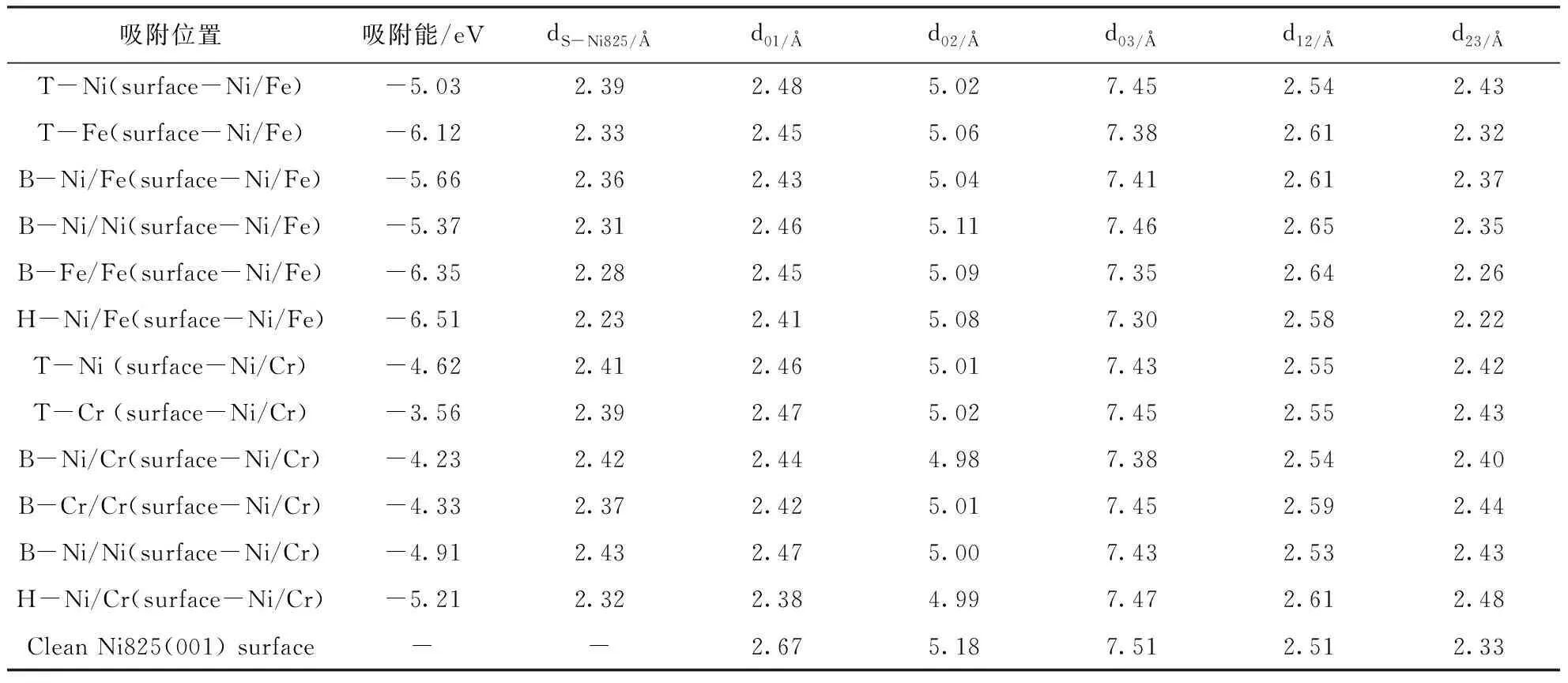
表4 0.25覆盖度下S在825合金(001)面各个高对称位置的吸附能和吸附后几何结构参数
表4中,dS-Ni825是S原子与825合金(001)表面之间的最短键长; d01, d02和d03分别表示在S吸附合金表面弛豫之后固定层与第一, 第二, 第三层之间的距离. d12, d23分别表示第一和第二层以及第二和第三层之间的距离.(固定层定义为第0层, 往上依次为第1层, 第2层和第3层)
吸附往往伴随着表面弛豫, 即原子间键长和表面层间距发生变化, 所以本文讨论了S在两个终止面各个位点吸附后与合金825(001)面原子之间的键长以及合金表面各层层间距的变化情况. 通过表4可以看出S原子在Ni/Fe终止面和Ni/Cr终止面各个高对称位置吸附后与合金825(001)面金属原子之间键长变化在2.28~2.41 Å之间, 吸附能变化范围在4.23~6.51 eV之间. S在Fe(001)面的吸附研究[14]表明, S在Fe(001)面吸附的吸附能在-7.62~-5.82 eV之间变化, S-Fe键长在1.22~1.98 Å之间变化; 而S在Fe(111)面的吸附研究也可以看出[10], S在Fe(111)面吸附的吸附能在-7.52~-6.93 eV之间变化, S-Fe键长在2.15~2.25 Å之间变化. 对比发现, S在合金825(001)面吸附的吸附能明显低于在Fe表面的吸附能, 在合金825(001)面吸附后S与表面金属原子的键长也较之更短, S在合金825(001)面的吸附作用弱于吸附Fe表面.
分析层间距弛豫数据可以发现, S原子与合金825(001)面发生了比较强烈的相互作用, 因此S原子吸附各个高对称位置优化后的层间距与清洁表面相比均发生了较为明显的变化. 分析Ni / Fe终止面S的吸附可以得到, H位的吸附导致0-1层, 0-2层和0-3层分别收缩了0.26 Å, 0.19 Å和0.21 Å;对于Ni / Fe-Bridge吸附, 相应的数据分别为0.24 Å(0-1层), 0.14 Å(0-2层)和0.10 Å(0-3层); 对于Ni-Top吸附, 它使0-1层收缩0.21 Å, 0-2层收缩0.17 Å, 0-3层收缩0.08 Å. 通过比较层间距的弛豫变化, 不难发现S吸附在H位引起的合金(001)面弛豫最大, 进一步说明原子S在825合金(001)面Ni / Fe终止面的H位吸附后与合金表面作用最强, Ni / Fe终止面的H位是S原子在合金825(001)面的最稳定吸附位. 通过比较S吸附导致弛豫后的d12和d23与清洁表面的d12和d23看出: 尽管合金表面三层原子整体收缩, 但层d12和d23之间的间距正在扩大, 表明吸附导致合金表面层间金属之间的键长增加, 即层间金属原子之间的相互作用减少.
3.2.2差分电荷密度
为了对S吸附合金825(001) 面后其间的电子相互作用进行更直观的分析, 我们计算了0.25ML覆盖度下S原子在Ni / Fe终止面H位和Ni / Fe终止面H位吸附后的差分电荷密度. 差分电荷密度的计算公式为[10]:
Δρ=ρS/825-Slab-ρ825-Slab-ρS/825-Slab
(2)
其中ρS/825-Slab,ρ825-Slab和ρS/825-Slab分别为体系的总电荷密度, 合金825(001)面平板模型的电荷密度, 吸附在合金825(001)面的S原子的电荷密度, 图中红色表示得电子, 蓝色表示失电子.

图3 0.25ML覆盖度下, S吸附在825合金(001)面Ni/Fe终止面和Ni/Cr终止面H位的差分电荷密度图: a) S在Ni/Fe终止面H位吸附;b)S在 Ni/Cr终止面H位吸附Fig. 3 Under 0.25ML coverage, the two-dimensional differential charge density map when S is adsorbed in H position on the 825 alloy (001) surface Ni/Fe-termination surface and Ni/Cr-termination surface a)S is adsorbed in H position on the 825 alloy (001) surface Ni/Fe-termination surface; b)S is adsorbed in H position on the 825 alloy (001) surface Ni/Cr-termination surface
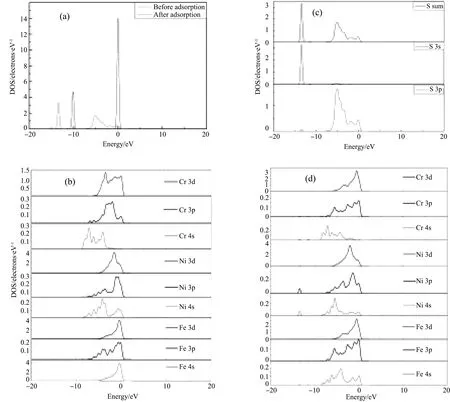
图4 S-合金825系统的总态密度和分波态密度 a)S在Ni / Fe终止面H位吸附前后的总态密度图; b)S吸附前清洁825合金(001)面的分波态密度图; c)S在Ni-Fe终止面H位吸附后S的分波态密度图; d)S在Ni-Fe终止面H位吸附后825合金(001)面的分波态密度图Fig.4 The total and partial density of states of S-825alloy system. a) DOS of S before and after S adsorption in H position on the 825 alloy (001) surface Ni/Fe-termination surface; b) DOS of Ni-based alloy 825(001) surface; c) PDOS of S after S adsorption in H position on the 825 alloy(001) surface Ni/Fe-termination surface; d) PDOS of Ni-based alloy 825(001) surface after S adsorption
电荷差分密度图能够直观明了地反映出S原子与合金825表面之间的相互作用所导致的电子重新分布状态. 由图3可以清晰得看出, S原子在Ni/Fe终止面H位和Ni/Cr终止面H位的吸附导致其与相邻最近四个金属原子发生了明显的相互作用, 他们的相互作用引起了电子之间的耦合, 杂化以及偏移. 无论是图3 a)还是3b)都能明显得看到, S的吸附导致合金825表面金属原子的电子密度主要集中在S原子的一侧, 使得S原子得到电子带负电, 基底的Fe原子和Ni原子失去电子带正电. 特别是S原子与Fe原子之间由于电子偏移所产生的耦合, 杂化使得S原子与Fe原子之间电子密度陡增而产生的红色区域颜色最深, 区域最大, 容易生成了腐蚀产物FexSy,与蔡晓文[16]的实验结果比较吻合. 然而, 通过图3b)我们发现, S的吸附并没导致基底表面Cr原子周围的电子密度发生明显变化, 说明Cr是825合金耐蚀性的关键添加元素. 比较图3 a)和3b)的电子密度变化色差也容易看出, S在Ni/Fe终止面H位的吸附时所引起的周围电子密度变化比在 Ni/Cr终止面H位吸附时大, 即S吸附在Ni/Fe终止面H位时较吸附在Ni/Cr终止面H位时与基底产生了更强烈的相互作用, 较好地印证了3.2.1中吸附能与表面弛豫的计算结果.
3.2.3电子态密度
为了进一步研究S吸附在合金825(100)面的电子结构, 计算了S原子在合金825(100)面Ni/Fe终止面H位吸附前后的电子态密度.
从图4a)的横坐标的能量分布可以看出, S原子吸附在Ni / Fe终止面H位后使得S原子的态密度峰个数增多, 由吸附前的2个增多到5个; 波峰的分布也具有向低能量区域移动的趋势, 波峰横坐标由吸附前的0 eV-1和-10 eV-1向左偏移到-5~0 eV-1和-15~-10 eV-1, 而且波峰峰值相比吸附前降低了很多, 从14左右降到约3~4, 说明S在Ni / Fe终止面H位的吸附使得合金基底表面原有的双电层结构发生变化,S与其表面产生了较为强烈的相互作用. 从b)和d)不难得到, Fe的3d和4s轨道的峰值, 特别是4s轨道峰由于S原子的吸附显著低于吸附前, 说明Fe的4s轨道上的电子向S偏移趋势明显, S原子的吸附容易导致FexSy腐蚀产物的生成, 这也与电荷差分密度图的计算结果相一致. 同样是在0.25 ML覆盖度下, 相比S原子的吸附导致纯Fe(001)面分波态密度的变化[14]程度(s,p,d轨道的波峰和波展均发生大的振幅), 从b)和d)图我们发现S原子的吸附导致镍基合金825(001)面中Fe的分波态密度变化却小得多(仅4s轨道变化明显), 说明合金825中的Cr和Ni对S与Fe之间的电荷相互作用有抑制作用. 另外, S吸附前后, 825合金基底Cr的态密度的变化也不大, 与电荷差分密度图符合得较好, 很好地解释了防腐合金生产中添加元素Cr能使合金具有较好耐蚀性. 结合图c)和图d)我们不难得到, 吸附在Ni / Fe终止面H位的S原子和825合金基底表面的相互作用主要是由S的3p轨道与Ni原子的3d轨道, Fe原子的4s轨道以及Fe原子的3d轨道耦合杂化贡献.
4 结 语
S原子在镍基合金825(001)面的最稳定吸附位为Ni / Fe终止面的四重穴位, 对应吸附能为-6.51 eV. S原子与825合金(001)面的相互作用主要是由S的3p轨道与Ni原子的3d轨道, Fe原子的4s轨道以及Fe原子的3d轨道耦合杂化贡献. 通过电荷差分密度图和态密度的电子表征可以发现, Fe与S之间电子杂化转移明显, 形成离子键, 较易生成腐蚀产物FexSy, 而S的吸附对合金中Cr原子的电子分布影响不大, 并且825合金中Cr和Ni的存在能很好地抑制S-Fe之间的相互作用, Cr是825合金具有较好耐蚀性的关键添加元素. 但本文只研究了0.25ML覆盖度下S在镍基合金825(001)面的微观吸附理论, 其他不同S覆盖度在合金另外两个低指数面(111)和(100)面的吸附有待进一步研究, 以完善S原子在镍基耐蚀合金825表面吸附的微观作用机理.
