三代测序与靶向捕获技术联用进行高分辨HLA基因分型及MHC区域单倍体型精细鉴定
2019-04-22陈佳舒明月里进付爱思杨帆王邹李一荣邓子新刘天罡
陈佳,舒明月,里进,付爱思,杨帆,王邹,李一荣, 邓子新,刘天罡
三代测序与靶向捕获技术联用进行高分辨基因分型及MHC区域单倍体型精细鉴定
陈佳1,舒明月1,里进2,付爱思1,杨帆3,王邹3,李一荣2, 邓子新1,刘天罡1
1. 武汉大学药学院,组合生物合成与新药发现教育部重点实验室,武汉 430071 2. 武汉大学中南医院检验医学中心,武汉 430071 3. 武汉生物技术研究院公共技术服务平台,武汉 430071
人类白细胞抗原(human leukocyte antigen,)的高分辨率、精准分型对于组织配型以及相关疾病研究具有重要意义。本研究以12位原发性肝细胞癌病人的外周血为供试样本,分析二、三代测序数据用于高分辨率分型的优劣势,同时结合探针捕获与三代测序技术对YH、HeLa标准细胞系以及一个原发性肝细胞癌病人的主要组织相容性复合体(major histocompatibility complex,MHC)区域进行靶向分析,探究长读长测序技术对于整个MHC区域精细分析的潜力。研究表明:(1)二、三代测序技术均能实现6~8位高分辨分型,且两者分型结果一致。但是三代数据的覆盖均一度显著优于二代,不会出现明显的“断层”现象;(2)超长的三代数据可直接跨越整个扩增子,对于基因单倍体型的判定(phasing)具有明显优势。样本中92.79%的基因能够得到准确的单倍体分型结果,远高于二代的75.65%;(3)长读长的三代测序数据不但能实现对MHC区域的更好组装,还具有对整个MHC共计3.6 Mb区域进行phasing的能力,而这将有助于明确各个突变位点、等位基因、非编码区等基因原件在每个MHC单倍体型上的定位与相互连锁信息,为免疫等相关疾病的研究提供理论依据。
;MHC;三代测序;单倍体分型;NimbleGen探针捕获技术
人类白细胞抗原(human leukocyte antigen,)基因位于人体第6号染色体的短臂,受控于人类主要组织相容性复合体(major histocompatibility complex, MHC)的基因簇[1],全长约3.6 Mb,是目前所知人体最复杂的遗传多态性系统[2,3]。
前期研究表明,基因的变异与传染病[4]、药物过敏反应[5]、自身免疫疾病[6]、器官移植反应[7]以及恶性肿瘤[1]等均有关联。此外,近期研究也表明特定等位基因多态性与原发性肝细胞癌的发生相关[8]。因此,准确的分型技术对于组织配型以及研究与疾病相关性具有重要意义。
由于MHC区域存在高度的多态性和广泛的连锁不平衡,因此研究人员对该区域所涉及到的分子机制的研究受到一定的限制。传统基于聚合酶链式反应等分型方法存在分辨率低、无法获得单倍体型结果以及新等位基因信息等诸多问题[9,10]。虽然以往Sanger测序技术被视为是分型的金标准[11],但其因通量低而逐步被各种第二代测序(next-generation sequencing, NGS)平台所取代[9,12,13]。但是,NGS对于个体所具备的两套同源染色体的独特核苷酸信息,即“对单倍体型的判定”(phasing)的解析依然存在困难,而已有文献表明准确的单倍体分型能更好解读基因与表型(包括疾病)之间的关系[14],尤其是对于等较大的基因,NGS因其读长短而很难准确获得单倍体型结果[15]。基于单分子实时测序技术(single molecule real-time, SMRT)的第三代测序仪能产生平均长度在10 kb以上的数据,这不但有利于基因精确分型,还能实现对基因组复杂区域的组装以及对某个基因内及等位基因间差异的细致解析。目前利用该技术进行基因组完成图组装或用于多倍体基因组中单倍体型的解析研究已有所报道[16,17]。
三代测序技术预期能实现对基因及MHC区域更精细的分析,从而准确地确定每个基因甚至整个MHC区域的单倍体分型结果,并且可有效挖掘包括单核苷酸多态性(single nucleotide polymorphhism, SNPs)在内的一系列遗传信息,这将极大地推进各类与人体免疫相关研究的发展。本研究以12位原发性肝细胞癌病人的外周血为供试样本,分析二、三代测序数据用于高分辨率分型的优劣势,同时结合探针捕获与三代测序技术对MHC区域进行靶向分析,探究长读长测序技术对于整个MHC区域精细分析的潜力。
1 材料与方法
1.1 研究对象
本研究收集的12位原发性肝细胞癌(hepaticellular carcinoma, HCC)患者外周血样本均由武汉大学中南医院提供,所有患者均签署了知情同意书。YH标准细胞系由中国国家基因库提供,HeLa细胞采购自美国菌种保藏中心。
1.2 HLA基因的测序分型
外周血样本总DNA提取使用Hipure Blood DNA Mini Kit (广州美基生物科技有限公司)完成,YH、HeLa细胞系总DNA的提取采用酚-氯仿提取法。基因扩增使用GENDX NGSgo-AmpX试剂盒以及QIAGEN Long range PCR试剂盒(QIAGEN公司,德国),扩增产物使用Qubit 2.0定量后等摩尔混合,不同样本的扩增子混合物分别采用Illumina Miseq以及PacBio RSII的标准混样建库流程进行文库制备及测序。PacBio RSII原始数据使用SMRT Portal中的RS_ReadsOfInsert方法进行质量过滤,得到环形一致性序列(circularCCS)将过滤的质量值(即minimum predicted accuracy参数)分别设为0.80、0.85、0.90、0.95和0.99,分别得到CCS0.80、CCS0.85、CCS0.90、CCS0.95和CCS0.99的高质量数据。
分型分析中使用的各种开源或商业分型软件均采用从官方网站或者商业公司处获得的最新版本,其中,NGSengine、HLAssign、HLA-reporter、Omixon、HLAminer、HLA-VBseq 和 OptiType用于二代数据分型,NGSengine和HLAminer用于三代不同数据类型以及不同质量值CCS的数据分型。
1.3 MHC区域捕获测序以及数据分析
使用Roche NimbleGen MHC探针捕获试剂盒对MHC相关区域(包括传统MHC区域和约1.0 Mb的MHC周边区域)进行捕获,实验过程根据三代测序长读长的特点对DNA打断、DNA纯化体系以及DNA杂交时间等操作进行了优化,采用PacBio RSII的标准流程进行文库制备及测序。使用FALCON软件进行数据组装并且使用SMRT Portal中RS_Resequencing标准流程将原始测序数据比对到对应参考基因组MHC参考序列,以计算数据覆盖度并根据原始覆盖度(该流程的默认参数)统计SNPs在MHC区域的分布(未对覆盖度进行筛选,95%以上的覆盖度为100×)。
MHC区域单体型分析分别采用了FALCON- Unzip软件以及targeted-phasing-consensus脚本(https://github.com/PacificBiosciences/targeted-phasing-consensus)两套方法,并使用MUMmer软件将得到的单倍体分型结果与其对应的基因分型结果进行比对,评估上述两套方法对MHC区域单倍体分型结果的差异。
2 结果与分析
2.1 基于二代测序数据对HLA基因分型
利用包括全基因组、全外显子组、转录组及基因扩增子数据在内的多种数据类型进行分型的各种学术和商业化软件已被广泛使用(表1)。为评估分型软件的准确性以便从中选择最佳的分型软件用于与后续三代数据分型结果的比较,比较了7种不同的分型软件对基于全长扩增子二代测序数据的分析性能。为了确保分型结果的可靠性,不论是Illumina Miseq还是PacBio RSII均保证了足够的数据量。由于二代数据存在一定偏好,因此其覆盖度会存在不均一的情况,不同基因的覆盖度有一定的差异(图1),但是总体而言,其95%以上的区域覆盖度会大于200×。而三代数据的覆盖度比二代数据更高(图1)。通过比较,7种软件对二代数据的分型结果展现出较大差异。从总体上来看,NGSengine对classⅠ和classⅡ基因的分型结果敏感度都较高,结果与血清学鉴定结果吻合。而其他6种分型软件仅对classⅠ类基因的分型均较为准确,但对classⅡ类基因则不敏感,部分软件甚至不能给出分型结果。例如,HLAssgin和HLAreporter只有2个(2/12)和0个样本(0/12)预测到了基因分型。而HLAminer和Omixon对classⅠ基因的分型结果的判定与NGSengine/血清学结果相比有很大的差异。由于HCC27样本的基因分型结果经过了血清学结果及MHC区域捕获测序结果的双重验证,故以该样本作为示例(表2),NGSengine对classⅠ和classⅡ基因的分型结果与血清学鉴定结果以及MHC区域捕获测序结果高度吻合。但是HLAssgin和HLAreporter无法预测到DPA1的基因分型,而HLAminer和Omixon对DPB1、DQB1、DRB1等classⅡ基因的分型结果判定错误率较高(表2)。
此外,OptiType与NGSengine的分型结果很相似,但是其分辨率只有4位(表2)。有文献表明,位于外显子外部的单核苷酸变异可能在疾病的发病机制中起关键作用[27]。因此,高分辨率的分型软件具有更高的应用价值。在测试的7种分型软件中,NGSengine分型最准确,分辨率最高。后续将以NGSengine产生的结果为参照,评估三代测序数据分析结果。
2.2 基于三代测序数据对HLA基因分型
采用两种已公开能够使用三代测序数据的分型软件——HLAminer和NGSengine对基于三代测序数据的HLA基因进行分型。此外,PacBio三代测序数据分为subreads和环形一致性序列(circularCCS)两类:subreads是去除接头序列和低质量部分所得到的未经矫正的数据,而CCS是将来自于同一个DNA分子经过环状反复测序产生的多条subreads相互矫正后得到的高准确性数据。理论上,CCS数据的准确性越高越有利于获得准确的分型结果,但是矫正准确性设置越高最终所获得的CCS数据量也会减少,存在不能满足分型最低数据量需求的风险。因此,本研究测试了不同的数据类型和CCS准确性对分型结果的影响。
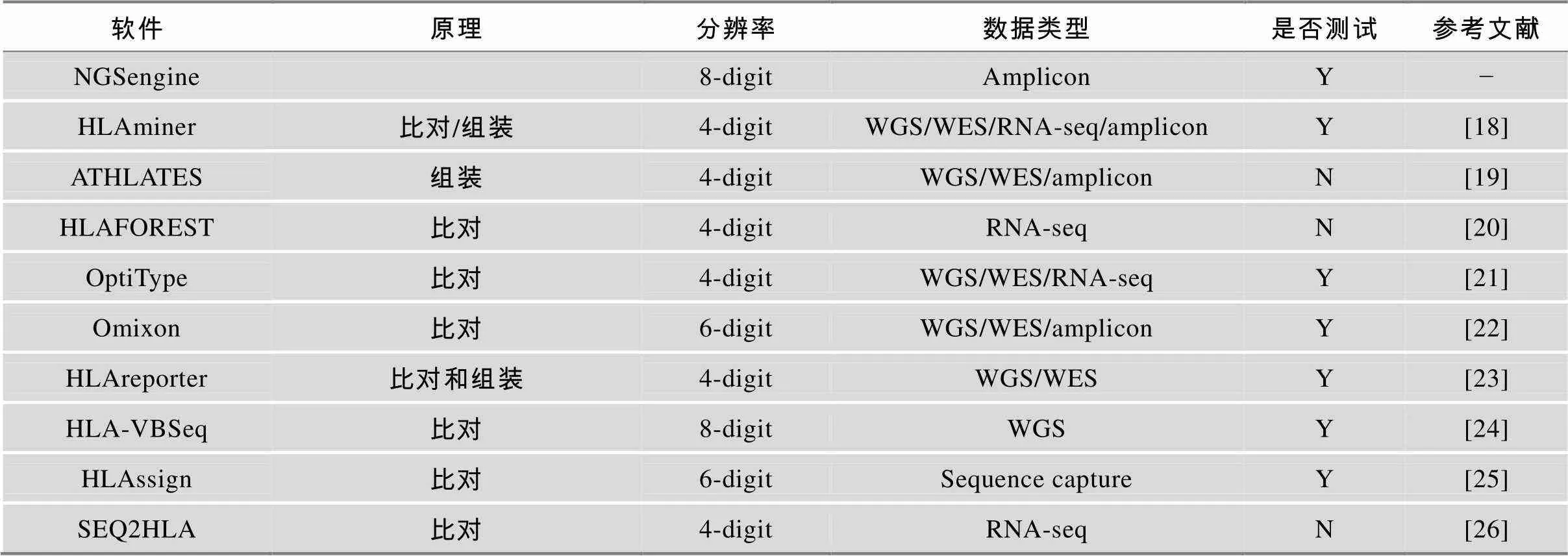
表1 7种HLA分型软件比较
WGS:全基因组数据;WES:全外显子组数据;RNA-seq:转录组数据;Amplicon:基因扩增子数据;Y:对应软件已测试;N:对应软件未测试。
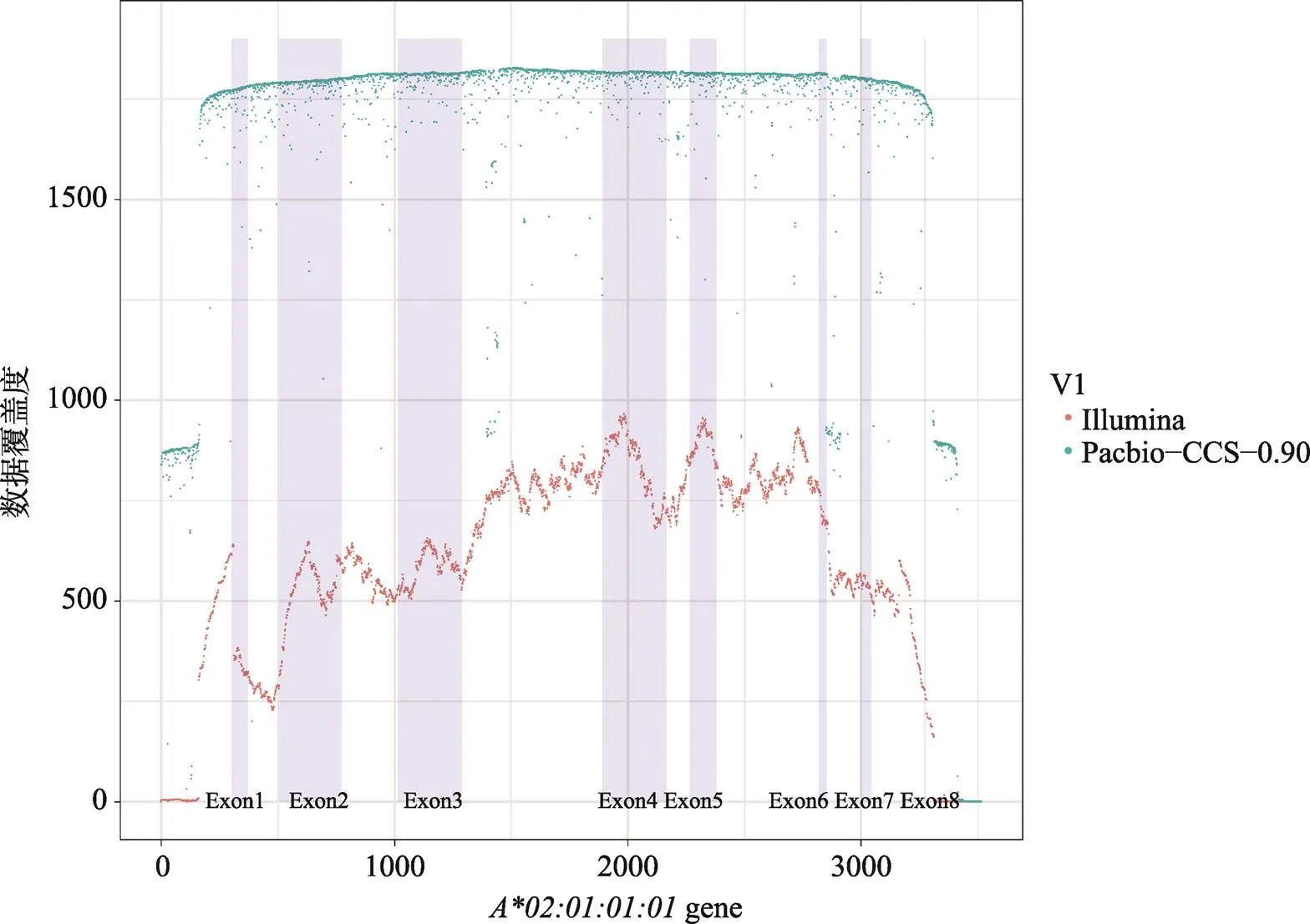
图1 HCC27样本HLA-A基因Illimina和PacBio-CCS-0.90数据覆盖度
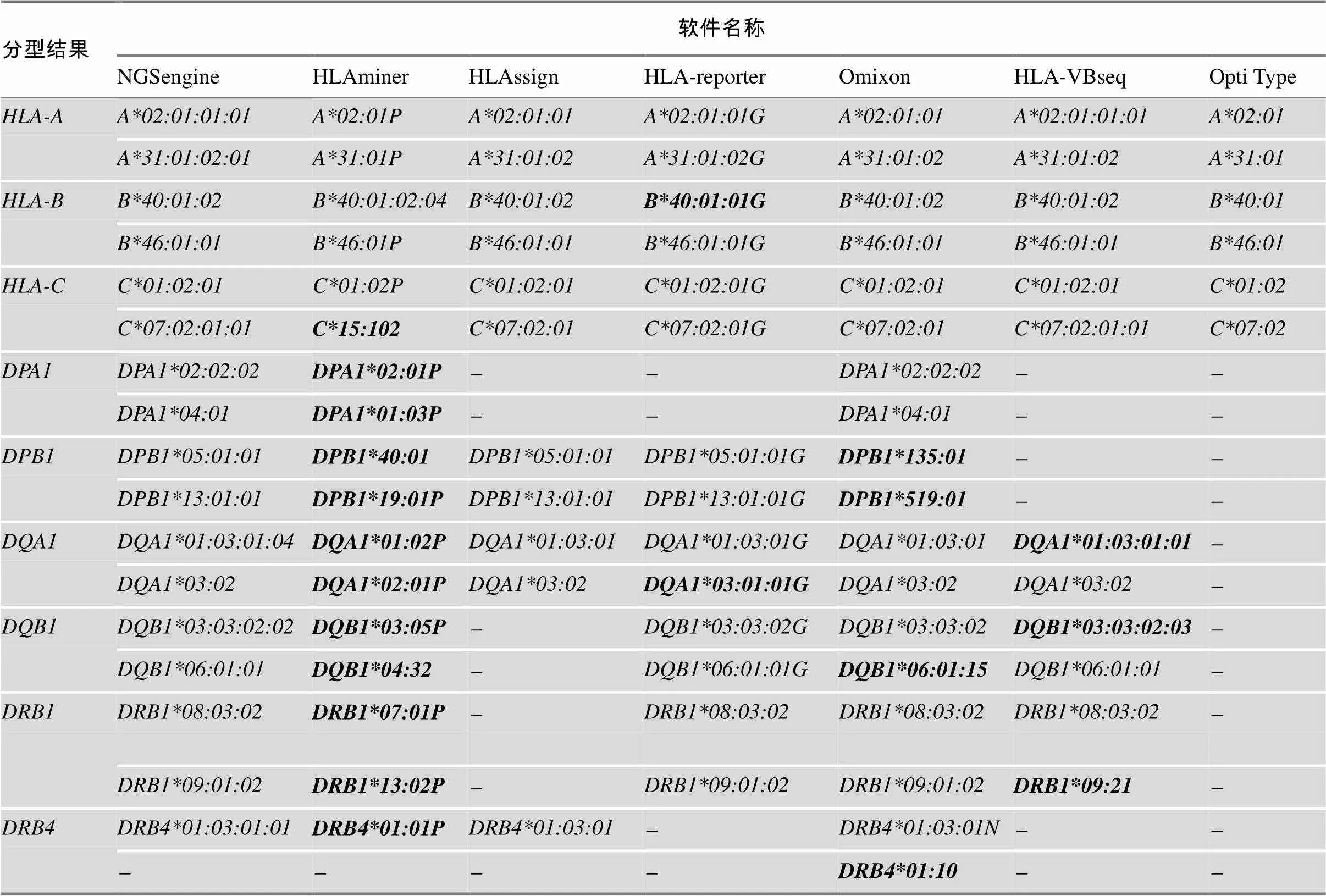
表2 HCC27样本基于二代测序数据的7种软件分型结果比较
加粗标记表明与NGSengine不一致的分型结果;“”表明无法得到该基因的分型结果。
首先,HLAminer软件三代与二代数据的分型结果间存在差异,而NGSengine软件三代与二代数据的分型结果保持一致(表3)。此外,当subreads的数据量足够时,基于subreads数据的分型结果与CCS数据基本一致,但前者往往会多出一个代表错配信息的后缀。以HCC5样本的基因为例,其基于subreads的分型结果为DRB5*02:02,而CCS对应的分型结果为,这表明subreads的结果在外显子区域存在3个错配碱基。从单碱基准确性角度考虑,使用CCS数据对基因分型应该是更好的选择。
随着CCS数据准确性的不断提高,其数据量会显著下降(图2)。以HCC27样本为例,数据量下降最大的两个断层分别在subreads到CCS0.80,以及CCS0.95到CCS0.99两处,其数据量分别降低了78.86%和89.84%。虽然CCS0.99准确性最高,但其reads数量从subreads的3000条降至50条,无法满足后续的分型需求。另一方面,数据量远多于CCS数据的subreads并没有获得最好的覆盖度,除了和,其他基因的subreads覆盖度甚至还低于CCS0.80 (图2),这可能是由于subreads错误率较高导致大量结果被过滤。因此,考虑到准确性和数据量之间的平衡,本研究最终选用CCS0.90的数据用于最终分型结果的比较。

表3 NGSengine和HLAminer的分型结果分辨率及与Illumina分型结果一致性统计
114个基因中有99个完全一致,15个不一致,分型结果体现在第8位分型结果的差异,这类差异可通过软件优化与参数调整进一步减少。
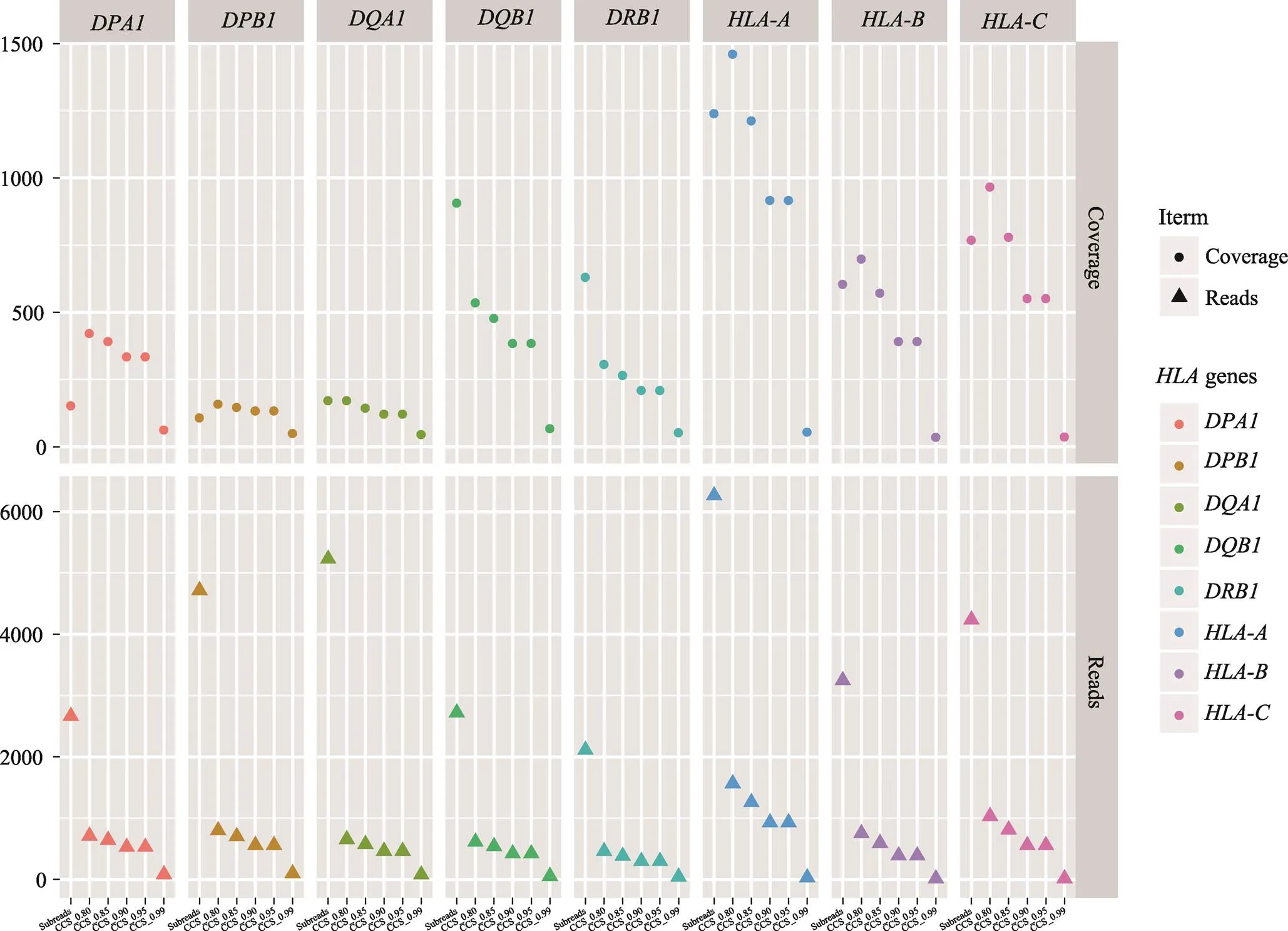
图2 HCC27样本在基于不同数据类型和CCS准确性下各HLA基因的覆盖度和数据量
虽然基于二代和三代CCS0.90数据的分型结果一致(表3),但是两种数据的基因覆盖度却存在较大差异(图1)。Illumina数据存在一定偏好,在某些区段上会出现明显的“断层”现象,尤其在基因两端区域。相比之下,CCS0.90数据的覆盖度更均匀,对于一些复杂的或全新的基因具有更强的分型能力和更低的错误风险。
2.3 基于三代测序数据对HLA基因进行单倍体分型
Phasing regions是用以评估单倍体分型效果的重要指标,主要代表目的基因中能准确分型单倍体区域的数目。分析结果显示基于二、三代数据进行基因单倍体分型的结果间存在一定的差异(表4)。基于三代数据,有92.79% (103/111)的基因可以得到一条完整的单体型结果,而这一比例在NGS数据里仅占75.65% (87/115)。与此同时,同一个单体型被定相到3个以上区域的比例中,NGS占比13.91% (16/115),而三代测序数据对应的占比仅为3.6% (4/111) (表4)。因此,三代测序更有利于提高单倍体分型的准确性,减少不确定性。
2.4 利用PacBio数据组装MHC区域以及SNP在MHC区域上的分布
MHC捕获探针设计区域大小为4 970 458 bp (Chr.6:28 477 797~33 448 354 bp),使用FALCON软件对YH标准细胞系进行组装,得到的最佳组装结果为:MHC组装大小为4.46 Mb,Contig N50为85 kb,Contig总数154个。将组装的Contig比对到YH基因组的参考序列上(图3),可以完整覆盖其MHC的参考序列。数据总覆盖度97.86% (4 864 179/4 970 458 bp),跟以往使用二代数据结果获得的覆盖度为97.29%的结果相比有所提升[28]。
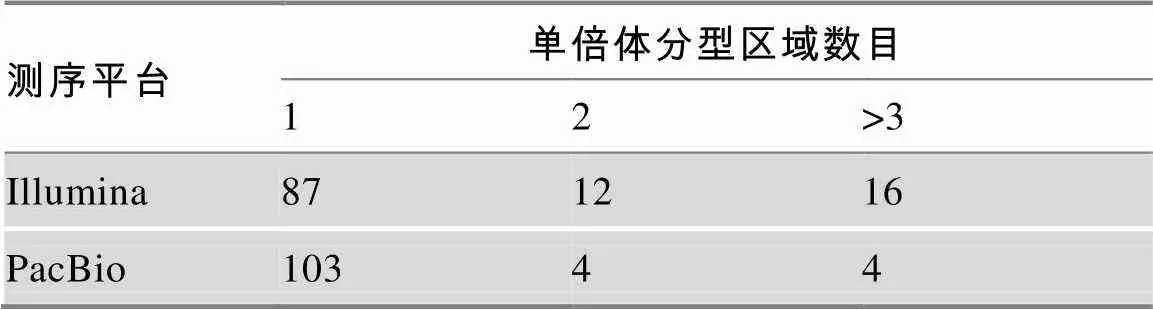
表4 二代(Illumina)和三代(PacBio)测序数据中所有HLA基因phasing regions个数的差异
随后,使用根据YH细胞系组装优化参数对HCC27样本MHC区域进行组装,得到的最佳组装效果为:MHC组装大小为4.79 Mb,Contig N50为90 kb,Contig总数223个。数据覆盖度99.8% (4 960 480 bp/ 4 970 458)。0×以下的覆盖度比例为0.21% (10 363/ 4 970 458 bp),30×以下的覆盖度比例为2.23% (110 974/4 970 458 bp),意味着有97.77%的序列覆盖度达到30×以上,组装效果进一步提升。
此外,本研究统计了HCC27样本、YH和HeLa细胞系MHC区域SNP的分布情况(图4),发现基因区域的SNP频率显著升高,该结果与之前报道的结果一致[28],这也是基因多态性高的重要表现。
2.5 关于HCC27样本MHC区域的单倍体分型
FALCON+FALCON-Unzip以及targeted-phasing- consensus采用了两种不同的单倍体分型原理。前者是基于数据的从头组装,是无参考序列的单倍体型分析方法。后者的分析思路是基于数据比对,将原始的测序数据比对到参考序列上,然后根据比对的结果得到两条单倍体型结果,即consensus0和consensus1。两种方法所得到结果与本文2.2部分的结果基本一致(表5)。为进一步验证上述HCC27样本基因单倍体分型结果的准确性,对其产生的两条单倍体型序列中所含有序列与各外显子区域内基因进行比对(图5,A和B),两者在单碱基水平上均保持一致。这表明基于长读长的三代测序数据,两种方法均可以对MHC区域上各基因进行较为准确的单倍体型分析。此外,基于捕获的测序结果,还可以获得扩增测序难以得到的内含子信息。
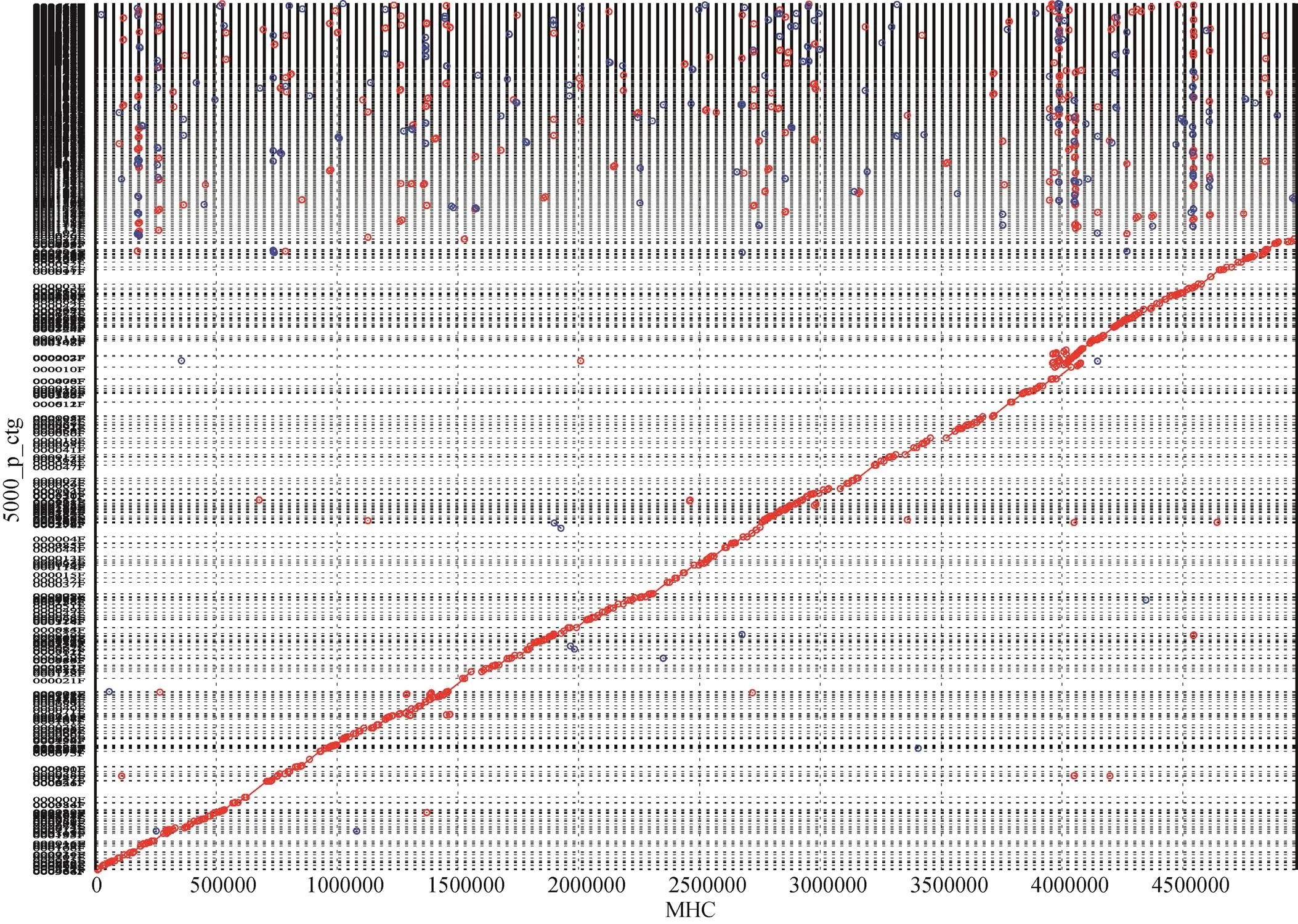
图3 YH细胞系MHC区域FALCOM组装序列与人类MHC的参考序列的比对结果
另一方面,FALCON+FALCON-Unzip会出现同一基因被比对到不同contig上的情况(表5),表明该组装方法对于MHC这类的多态性极高的区域可能会存在一定的单倍体分型错误。而基于比对方法得到的consensus序列,由于捕获测序长度限制,也可能导致在进行单倍体分型时,同一个位点的SNP信息难以准确定位到不同的consensus序列,从而导致模棱两可的结果。
因此,本研究整合了上述两种单倍体分型方法的所有信息以及各基因扩增子的分型信息,对HCC27样本MHC区域的单倍体上的基因的分布进行了校正和预测(图6),两套基因分别被定位到consensus0和consensus1两个单倍体型上。其中,没有用虚线标注的,即和等位基因,还通过从头组装的方法(FALCON+FALCON- Unzip结果)验证了这两个等位基因的确位于一条contig上。基于此,可以大致了解来自双亲的两套等位基因、以及基因间其他功能原件在MHC上的确切位置与连锁关系,这对更深入研究基因与表型(包括疾病)之间的关系具有重要意义。
3 讨论
本研究评估了7种可使用二代测序数据和2种可使用三代数据的分型软件。结果显示,二代数据的分型结果差异很大,其主要归因于各软件最适合输入数据类型、分析原理、数据库的差异(表1)。而不论是基于二代还是三代数据,NGSengine均能产生准确,分辨率高的分型结果,这说明分型实验设计与分析流程匹配的重要性。CCS数据有助于提高分型结果的单碱基准确性,基于CCS分型结果其外显子错配信息会大幅减少,需要进行单碱基级别分析的研究可采用CCS0.90的数据进行分析。不同于二代数据需要通过多条数据组装/计算等分析手段对基因上各个位点的相位进行单倍体分型。单条三代测序数据可跨越较长的区域,基因分型与单倍体分型过程不涉及数据组装,减少了因组装而导致的错误。虽然在本研究中二、三代的分型结果基本一致,但是三代的覆盖均一度和单倍体分型结果均优于二代(图1,表4),可以大幅提高单倍体分型的准确性,减少模棱两可的分型结果,更适用于基因的分型与单体型分析。
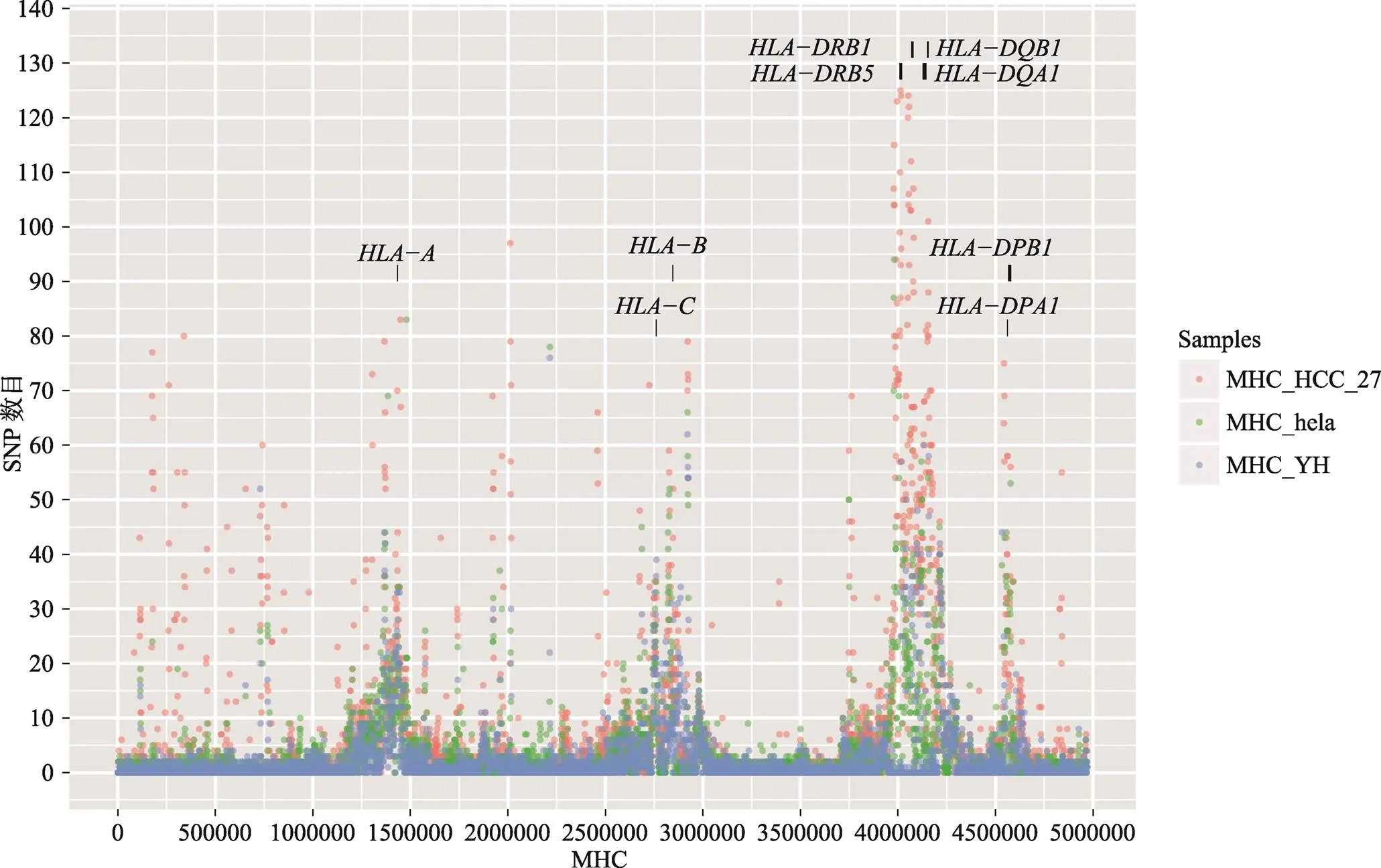
图4 HCC27、YH和HeLa标准细胞系SNP在MHC区域的分布图
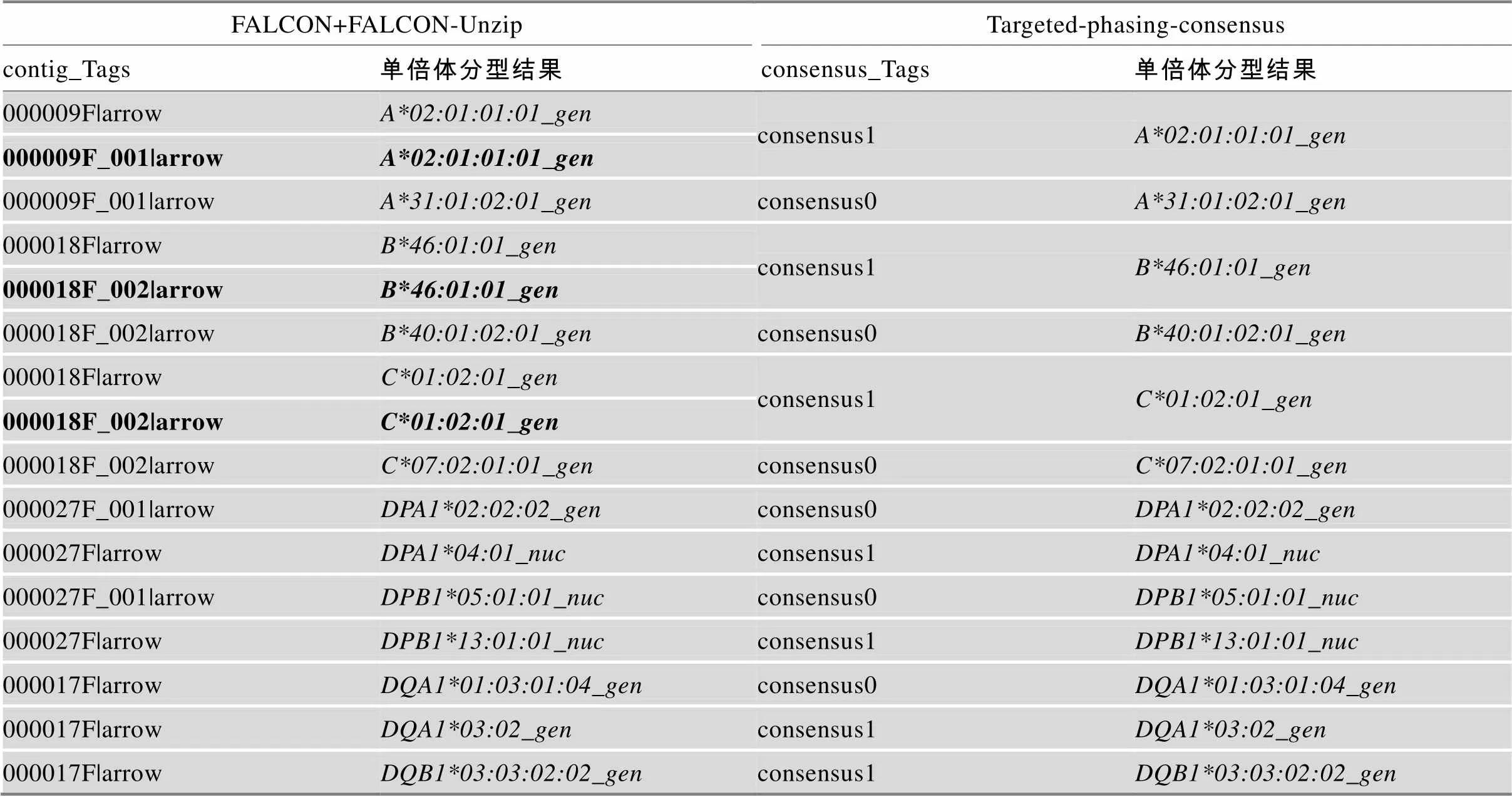
表5 HCC27基于FALCON+FALCON-Unzip以及Targeted-phasing-consensus的单倍体分型结果
contig_Tags和consensus_Tags表明单倍体分型结果对应的contig和consensus编号;加粗标记表示同一单倍体分型被比对到不同的contig上。
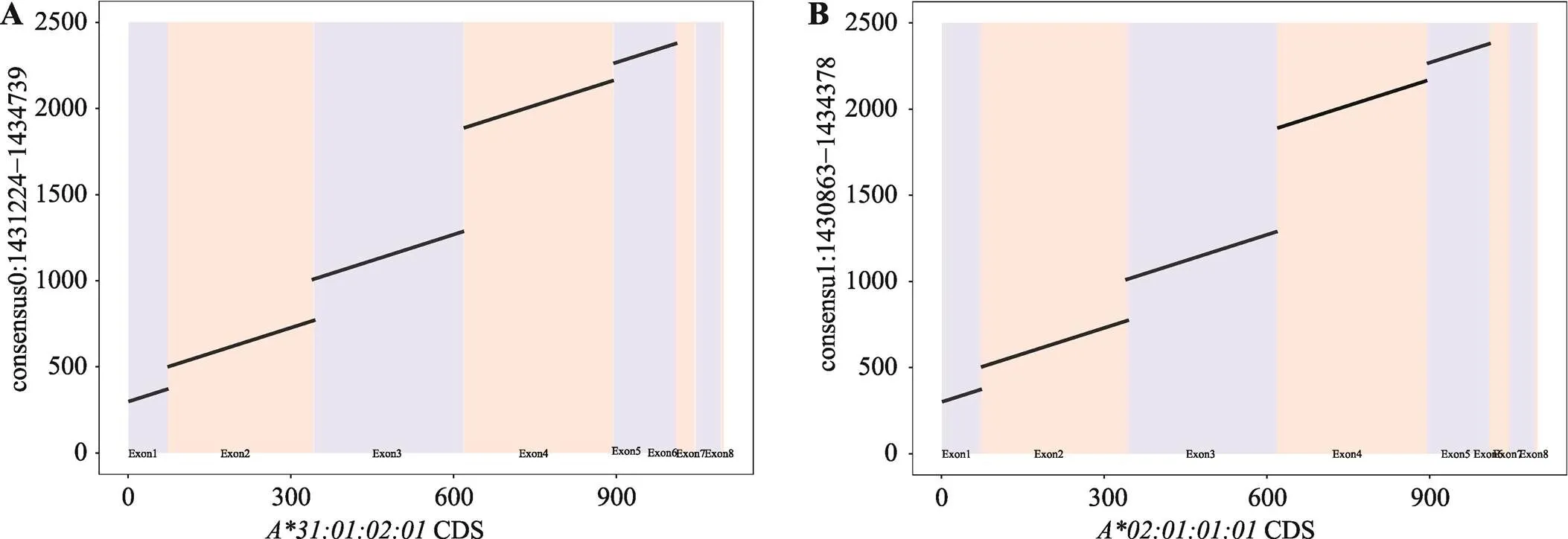
图5 HCC27样本consensus0和consensus1与HLA-A基因的比对结果
A:HCC27样本consensus0与的序列比对结果;B:HCC27样本consensus1与的序列比对结果。
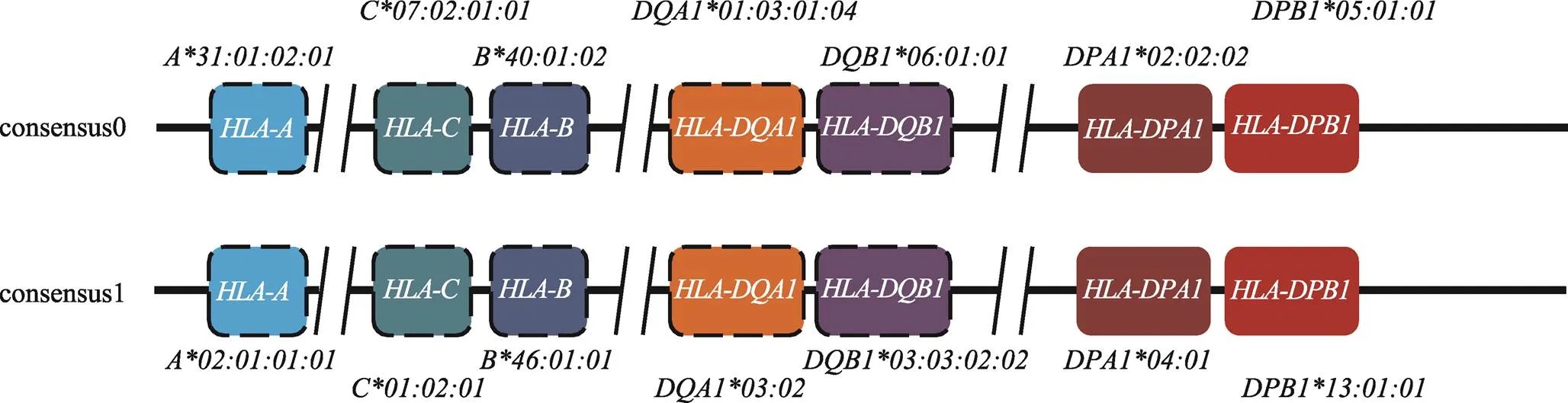
图6 HCC27 MHC区域的单倍体分型结果预测
consensus0和consensus1表示利用targeted-phasing-consensus脚本得到的两个单倍体型,不同颜色表示不同的等位基因。未用虚线标注的,表明其结果可通过从头组装的结果(FALCON+FALCON-Unzip结果)得到验证,虚线边框标注的表示通过从头组装结果无法得到验证信息。
YH、HeLa和HCC样本的MHC区域捕获和三代测序的结果表明,三代数据的组装结果优于以往文献使用二代测序的结果,且结果准确性可以达到单碱基水平。此外,FALCON+FALCON-Unzip软件由于是基于三代测序原始数据的无参考基因组单倍体分型方式,可能出现同一基因被比对到不同Contig上的情况(表5),从而导致组装出错。而targeted- phasing-consensus方法虽然是基于参考基因组序列的单倍体分型方法,但由于受到捕获产物测序数据长度的限制,同一个位点的SNP位点难以准确定位到不同的consensus序列,同样可能导致模棱两可的结果。因此,本研究将上述两种基于组装和比对的单倍体分型方法所得到的单倍体型信息以及本文2.2部分的基因扩增子分型结果进行整合,对HCC27样本MHC区域的单倍体上的基因的分布进行了校正和预测,通过从头组装的方法对预测结果进行验证,发现和等位基因的确位于同一contig上。基于该方法,可以从整体上了解来自于双亲的两套等位基因、以及基因间其他功能原件在MHC上的位置与连锁关系,这将有助于对MHC这类结构复杂的基因区域进行系统研究并极大的推进各类相关疾病的相关性分析。
[1] TANG MZ, CAI YL, ZHENG YM, ZENG Y. Association between human leukocyte antigenand nasopharyngeal- carcinoma., 2012, 34(12): 1505–1512.汤敏中, 蔡永林, 郑裕明, 曾毅. 人类白细胞抗原与鼻咽癌的相关性. 遗传, 2012, 34(12): 1505–1512.
[2] YANG Zhao-Qing, CHU Jia-You. The research progress of human genetic diversity in China., 2012, 34(11): 1351–1364.杨昭庆,褚嘉祐. 中国人类遗传多样性研究进展. 遗传, 2012, 34(11): 1351–1364.
[3] XU Jun-Pin, DENG Zhi-Hui, JU Gong-Yan, GAO Su-Jing, WANG Da-Meng, HE Liu-Mei, WEI Tian-Chi. Cloning and sequencingandgenomic DNA and analyzing polymorphism in regulatory regions in Chinese Han individuals., 2010, 32(7): 685–693.徐筠娉,邓志辉,邹红岩,高素青,王大明,何柳媚,魏天莉. 中国汉族个体基因全长序列的测定及调控区多态性. 遗传, 2010, 32(7): 685–693.
[4] Kløverpris HN, Adland E, Koyanagi M, Stryhn A, Harndahl M, Matthews PC, Shapiro R, Walker BD, Ndung'u T, Brander C, Takiguchi M, Buus S, Goulder P. HIV subtype influences HLA-B*07:02-associated HIV disease outcome., 2014, 30(5): 468–475.
[5] Mallal S, Nolan D, Witt C, Masel G, Martin A, Moore C, Sayer D, Castley A, Mamotte C, Maxwell D, James I, Christiansen FT. Association between presence of HLA-B* 5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir., 2002, 359(9308): 727–732.
[6] Galeazzi M, Sebastiani GD, Passiu G, Angelini G, Delfino L, Asherson RA, Khamashta MA, Hughes GR. HLA-DP genotyping in patients with systemic lupus erythematosus: correlations with autoantibody subsets., 1992, 19(1): 42–46.
[7] Sasazuki T, Juji T, Morishima Y, Kinukawa N, Kashiwabara H, Inoko H, Yoshida T, Kimura A, Akaza T, Kamikawaji N, Kodera Y, Takaku F. Effect of matching of class I HLA alleles on clinical outcome after transplantation of hematopoietic stem cells from an unrelated donor. Japan Marrow Donor Program., 1998, 339(17): 1177–1185.
[8] Donaldson PT, Ho S, Williams R, Johnson PJ. HLA class II alleles in Chinese patients with hepatocellular carcinoma., 2001, 21(2): 143–148.
[9] Lind C, Ferriola D, Mackiewicz K, Heron S, Rogers M, Slavich L, Walker R, Hsiao T, McLaughlin L, D'Arcy M, Gai X, Goodridge D, Sayer D, Monos D. Next-generation sequencing: the solution for high-resolution, unambiguous human leukocyte antigen typing., 2010, 71(10): 1033–1042.
[10] Shiina T, Suzuki S, Ozaki Y, Taira H, Kikkawa E, Shigenari A, Oka A, Umemura T, Joshita S, Takahashi O, Hayashi Y, Paumen M, Katsuyama Y, Mitsunaga S, Ota M, Kulski JK, Inoko H. Super high resolution for single molecule-sequence-based typing of classical HLA loci at the 8-digit level using next generation sequencers., 2012, 80(4): 305–316.
[11] Latham K, Little AM, Madrigal JA. An overview of HLA typing for hematopoietic stem cell transplantation., 2014, 1109: 73.
[12] Hosomichi K, Jinam TA, Mitsunaga S, Nakaoka H, Inoue I. Phase-defined complete sequencing of the HLA genes by next-generation sequencing., 2013, 14: 355.
[13] Barone JC, Saito K, Beutner K, Campo M, Dong W, Goswami CP, Johnson ES, Wang ZX, Hsu S. HLA-genotyping of clinical specimens using Ion Torrent-based NGS., 2015, 76(12): 903–909.
[14] Tewhey R, Bansal V, Torkamani A, Topol EJ, Schork NJ. The importance of phase information for human genomics., 2011, 12(3): 215–223.
[15] Nelson WC, Pyo CW, Vogan D, Wang R, Pyon YS, Hennessey C, Smith A, Pereira S, Ishitani A, Geraghty DE. An integrated genotyping approach for HLA and other complex genetic systems., 2015, 76(12): 928–938.
[16] Chin CS, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, Clum A, Copeland A, Huddleston J, Eichler EE, Turner SW, Korlach J. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data., 2013, 10(6):563–569.
[17] Bowman B, Ranade S, Harting J, Lleras, R. A novel analytical pipeline for de novo haplotype phasing and amplicon analysis using SMRT® sequencing technology., 2014, 25(Suppl.): S17–S18.
[18] Warren RL, Choe G, Freeman DJ, Castellarin M, Munro S, Moore R, Holt RA. Derivation of HLA types from shotgun sequence datasets., 2012, 4(12): 95.
[19] Liu C, Yang X, Duffy B, Mohanakumar T, Mitra RD, Zody MC, Pfeifer JD. ATHLATES: accurate typing of human leukocyte antigen through exome sequencing., 2013, 41(14): e142.
[20] Kim HJ, Pourmand N. HLA typing from RNA-seq Data Using Hierarchical Read Weighting., 2013, 8(6): e67885.
[21] Szolek A, Schubert B, Mohr C, Sturm M, Feldhahn M, Kohlbacher O. OptiType: precision HLA typing from next-generation sequencing data., 2014, 30(23): 3310–3316.
[22] Major E, Rigó K, Hague T, Bérces A, Juhos S. HLA typing from 1000 genomes whole genome and whole exome illumina data., 2013, 8(11): e78410.
[23] Huang Y, Yang J, Ying D, Zhang Y, Shotelersuk V, Hirankarn N, Sham PC, Lau YL, Yang W. HLAreporter: a tool for HLA typing from next generation sequencing data., 2015, 7(1): 25.
[24] Nariai N, Kojima K, Saito S, Mimori T, Sato Y, Kawai Y, Yamaguchi-Kabata Y, Yasuda J, Nagasaki M. HLA-VBSeq: accurate HLA typing at full resolution from whole-genome sequencing data., 2015, 16(S2): S7.
[25] Wittig M, Anmarkrud JA, Kässens JC, Koch S, Forster M, Ellinghaus E, Hov JR, Sauer S, Schimmler M, Ziemann M, Görg S, Jacob F, Karlsen TH, Franke A. Development of a high-resolution NGS-based HLA-typing and analysis pipeline., 2015, 43(11): e70.
[26] Boegel S, Löwer M, Schäfer M, Bukur T, de Graaf J, Boisguérin V, Türeci O, Diken M, Castle JC, Sahin U. HLA typing from RNA-Seq sequence reads., 2012, 4(12): 102.
[27] Hosomichi K, Shiina T, Tajima A, Inoue I. The impact of next-generation sequencing technologies on HLA research., 2015, 60(11): 665–673.
[28] Cao H, Wu J, Wang Y, Jiang H, Zhang T, Liu X, Xu Y, Liang D, Gao P, Sun Y, Gifford B, D'Ascenzo M, Liu X, Tellier LC, Yang F, Tong X, Chen D, Zheng J, Li W, Richmond T, Xu X, Wang J, Li Y. An integrated tool to study MHC region: accurate SNV detection and HLA genestyping in human MHC region using targeted high-throughput sequencing., 2013, 8(7): e69388.
The third-generation sequencing combined with targeted capture technology for high-resolutiontyping and MHC region haplotype identification
Jia Chen1, Mingyue Shu1, Jin Li2, Aisi Fu1, Fan Yang3, Zou Wang3, Yirong Li2, Zixin Deng1, Tiangang Liu1
The high-resolution and accurate typing of human leukocyte antigen (HLA) is of great significance for the study of tissue matching in organ transplantation and the correlation betweenand disease. In this study, the peripheral blood of 12 patients with primary hepatocellular carcinoma was used to compare the advantages and disadvantages of the next- and third-generation sequencing technology for high-resolutiontyping. In addition, probe capture technology was used to capture the MHC region of YH and HeLa standard cell lines, and a primary hepatocellular carcinoma patient. The captured products were sequenced using PacBio platform to assess the potential of ultra-long reads sequencing technology for analysis of the entire MHC region. Our results showed that: (1) the next- and third-generation sequencing technology can both achieve 6-8 digit high resolution intyping. However, the coverage of the third-generation is significantly better than the next-generation sequencing technology. (2) The ultra-long reads of the third generation sequencing can directly span the entire amplicon region, which has obvious advantages for haplotype phasing, with 92.79% of thegenes having accurate phasing results, which is much higher than the 75.65% from the next-generation data. (3) The long-reads from the third generating sequencing can not only be used to assemble the MHC region but also the ability to phase the entire MHC region of 3.6 Mb, thereby helping to clarify the localization information of the mutation sites, alleles and non-coding regions on each MHC haplotype, and providing a theoretical basis for the study of immune and other related diseases.
; MHC; the third-generation sequencing;haplotype phasing; NimbleGen probe capture technology
2018-11-26;
2019-01-25
“万人计划”青年拔尖人才项目资助[Supported by the Young Talents Program of National High-level Personnel of Special Support Program (The “Ten Thousand Talent Program”)]
陈佳,在读硕士研究生,专业方向:微生物与生化制药。E-mail: chenjia19940216@whu.edu.cn
李一荣,主任医师,研究方向:临床分子免疫学诊断。E-mail: liyirong838@163.com邓子新,教授,研究方向:合成生物学。E-mail: zxdeng@whu.edu.cn刘天罡,教授,研究方向:合成生物学。E-mail: liutg@whu.edu.cn
10.16288/j.yczz.18-282
2019/2/25 17:19:18
URI: http://kns.cnki.net/kcms/detail/11.1913.R.20190225.1719.008.html
(责任编委: 方向东)
